
Qingying FENG, Dong LIU, Ying ZHANG, Hao FENG, Qiang LI. Thermodynamic and First-principles Assessments of Materials for Solar-driven CO2 Splitting Using Two-step Thermochemical Cycles [J]. Journal of Inorganic Materials, 2022, 37(2): 223
- Journal of Inorganic Materials
- Vol. 37, Issue 2, 223 (2022)
Abstract
Using solar energy to convert carbon dioxide (CO2) into carbon-neutral fuels provides an alternative to fossil fuels and mitigates global warming. In analogy to conventional chemical plants, a solar-fuel plant is able to operate overnight addressing the intermittency of solar energy. Regarding this, concentrated solar thermochemical CO2 splitting holds promise because cost-effective and high-density heat storage can be integrated into this technology[1]. Direct CO2 splitting by thermolysis, requires temperature as high as 3000 ℃ except for separation of oxygen from fuel products. Therefore, research converged to the two-step redox cycles based on partial reduction (or non-stoichiometric, oxygen-vacancy) and oxidation of non-volatile oxides (or redox materials), because this type of cycles features the combinations of practical operation temperature and high thermodynamic efficiency[1,2,3].
$\text{C}{{\text{O}}_{2}}\to \text{CO}+\frac{1}{2}{{\text{O}}_{2}}$, at Tth (Reaction (1), CO2 thermolysis)
$\frac{1}{\delta }\text{M}{{\text{O}}_{x}}\to \frac{1}{\delta }\text{M}{{\text{O}}_{x-\delta }}+\frac{1}{2}{{\text{O}}_{2}}$, at TH (Reaction (2), thermal reduction)
$\frac{1}{\delta }\text{M}{{\text{O}}_{x-\delta }}\text{+C}{{\text{O}}_{2}}\to \frac{1}{\delta }\text{M}{{\text{O}}_{x}}+\text{CO}$, at TL (Reaction (3), CO2 splitting)
Although considerable materials[4] have been examined including ceria[5,6,7,8], ferrites[9,10,11] and perovskites[12,13,14], the state-of-the-art solar-to-chemical energy conversion efficiency was as low as ~5% for solar thermochemical CO2 splitting[5]. Therefore, computational assessments of materials are still of great importance[15,16,17]. Intuitively, the thermodynamically suitable redox materials can be identified based on the fundamental concept that the change in the Gibbs free energy, ΔG, should be negative for the two cycle steps (Reactions (2) and (3)). Thereby, solid-state enthalpy and entropy of reduction excluding the contributions from gaseous species (whose thermodynamic property data is available for a wide range of conditions) can be used as the descriptors for thermodynamic assessments of redox materials. Particularly, Meredig[18] and Shah, et al.[19] raised the importance of entropic effects. Unfortunately, obtaining reliable thermodynamic data, especially the reduction entropy, of redox material candidates, either computationally or experimentally is often challenging[11-12,20-24]. For example, Bork, et al.[12] used CALPHAD models to access or extrapolate needed thermodynamic properties of perovskites, but models with this level of sophistication are not available for many redox material candidates[2]. Michalsky, et al.[22] found that the Gibbs free energy for formation of many bulk oxides (equivalent to solid-state enthalpy and entropy of reduction) scaled with their oxygen-vacancy formation energy at their most stable surfaces. However, this scaling feature was only demonstrated in the stoichiometric regime. Muhich, et al.[11] developed an assessment method where only the enthalpy of reduction was used, but this method only worked for the assessments within the same class of materials (hercynite in their work) because the reduction entropy is approximately the same for the hercynite family.
Therefore, this study provided general descriptors and criteria for thermodynamic assessments of proposed or newly discovered redox materials, and to provide first- principles methods for fast predictions of these descriptors. Here, pure and Samaria-doped ceria were taken as benchmark materials because they own fast splitting kinetics, excellent high-temperature and cycling stability[5,6,7,8], and also provide a platform for first-principles investigations of the effects of typical defects including oxygen vacancies, polarons and dopant ions[25].
1 Thermodynamic derivation of descriptors and criteria
In this section, the descriptors was proposed, and then the criteria was derived for thermodynamic assessments of the viability of material candidates. In the derivation, complementary constraints were presented in consideration of non-stoichiometric reaction mechanism, theoretical efficiency and practical operating conditions of the solar- driven two-step thermochemical process to extend the thermodynamic framework described by Meredig and Wolvertor[18].
This study starts with the fundamental concept that the change in the Gibbs free energy, ΔG, should be negative for the two cycle steps (Reactions (2) and (3)) so that they are thermodynamically favorable. This gives Eq. (1-2).
where ΔHf represents the enthalpy of formation, S represents entropy, P represents pressure (kPa) and R is the ideal gas constant. Solid-state entropy and solid-state enthalpy of formation are assumed to be temperature- independent. Observed from Eq. (1) and (2), solid-state change of entropy, ΔSsolid, and change of enthalpy of formation, ΔHsolid
depend on the redox material, while the rest quantities for gaseous species are independent and were well documented[26]. Therefore, ΔSsolid with unit of J∙(0.5 mol O2)-1∙K-1 and ΔHsolid with unit of kJ∙(0.5 mol O2)-1, defined in Eq. (3), were used as descriptors for thermodynamic assessments in this work. By substituting Eq. (3) into Eq. (1) and (2), Eq. (4-5), two of the assessment criteria, are obtained.
In the next step, thermodynamic principles are applied to analyse the solar-to-chemical (STC) energy conversion efficiency of the two-step thermochemical process to provide complementary criteria. STC efficiency, ηSTC = ηSTT×ηTTC, where ηSTT is solar-to-thermal efficiency, and ηTTC is thermal-to-chemical efficiency. Ideally, ηTTC=ΔG1298K/ ΔHsolid, where ΔG1298K (~256 kJ∙(0.5 mol O2)-1) is the change in the Gibbs free energy at 298 K for CO2 thermolysis (Reaction 1). By assuming an achievable ηSTT of 70%, a target ηTTC of 28.6% is required to achieve a practical ηSTC of 20%, which is competitive to the technology that a solar photovoltaic device coupled with an electrolyser[27]. Miller, et al.[2] analysed that potential redox materials should be able to achieve a theoretical thermal-to-chemical efficiency which is twice of the target ηTTC (Eq. (6)).
Therefore, Eq. (4) to (6) were used to calculate the boundary values of (ΔSsolid, ΔHsolid). Eq. (4) and (5) tell that the (ΔSsolid, ΔHsolid) boundary varies with operating temperatures (TH and TL) and operating pressure (PO2). For the thermal reduction step of a practical solar thermochemical reactor, TH=2000 K[18] and PO2=0.101 kPa[2] were used as the upper limit for the reduction temperature and the lower limit for the pressure respectively. For the oxidation step, the exothermic reaction is thermodynamically favorable for low temperatures, but suffers from slow kinetics. Thus, TL of 1000 K[18] was used as the lower limit for the oxidation temperature. Now, the criteria is summarized as Eq. (7) with the descriptors, ΔSsolid and ΔHsolid, defined in Eq. (3).
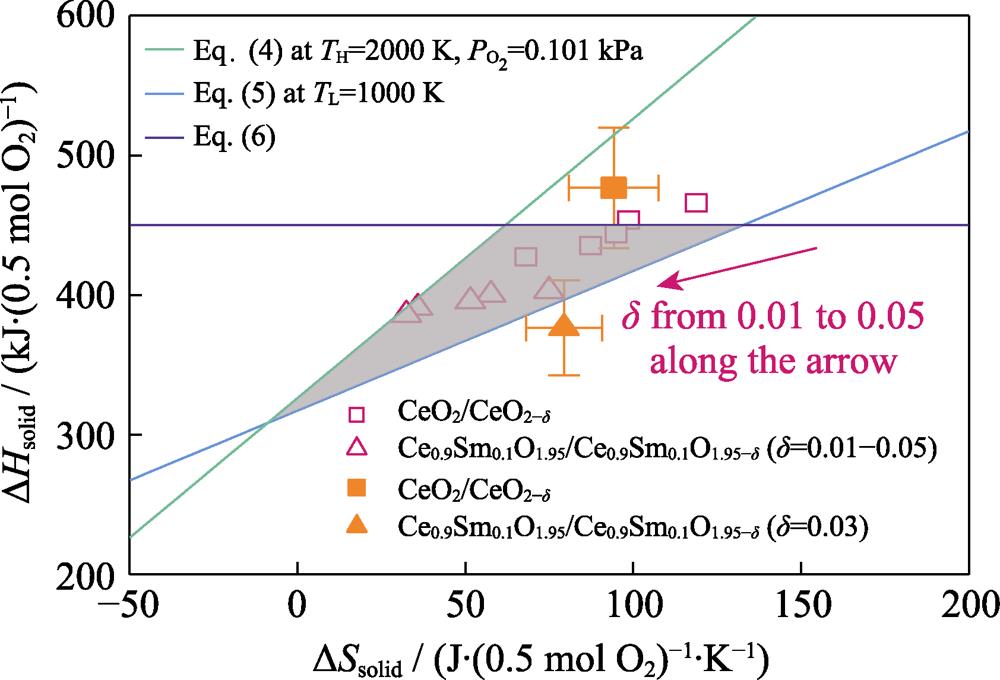
As shown in Fig. 1, the (ΔSsolid, ΔHsolid) area below the purple curve features theoretical thermal-to-chemical efficiency which is twice higher than the target. (ΔSsolid, ΔHsolid) below the green curve and above the blue curve are thermodynamically favorable for thermal reduction step (Reaction (2)) and CO2 splitting step (Reaction (3)) respectively. Therefore, the combination of ΔSsolid and ΔHsolid of qualified redox material should fall in the shaded triangular region.
![]()
Figure 1.Thermodynamic assessment mapThe combination of Δ
These results highlight the importance of accurate predictions of ΔSsolid and ΔHsolid, which are investigated in Section 2. Fig. 1 also shows that a positive ΔSsolid opens the favorable region, so redox materials with large positive ΔSsolid benefits reaction thermodynamics. However, this is non-trivial for redox materials in the stoichiometric regime, because the reduced oxide has fewer atoms and thereby, fewer vibrational degrees of freedom.
Fig. 1 plots the experimental data[28,29] of (ΔSsolid, ΔHsolid) for pure CeO2/CeO2-δ and 10% Samaria-doped Ce0.9Sm0.1O1.95/Ce0.9Sm0.1O1.95-δ redox pairs (δ=0.01-0.05) as well. Results show that ΔSsolid, ΔHsolid of both pure and doped ceria fall in the favorable region except for the pure ceria redox pair with δ=0.01. These results are reasonable because pure and doped ceria are usually used as benchmark redox materials in literature[5,6,7,8].
The favorable region varies with operating temperature and pressure as shown in Fig. 2. Therefore, it is important to recognize that the developed thermodynamic framework can further be used for reactor testing to design optimal operating condition of a specific qualified redox pair. For example, results show that thermodynamically favorable thermal reduction step (Reaction (2)) can be performed at lower temperatures (Fig. 2(a)) and higher pressures (Fig. 2(b)) for pure and doped ceria redox pairs with larger ΔSsolid (smaller δ). These milder operating conditions can significantly reduce thermal radiation loss and reactor design complexity.
![]()
Figure 2.Variations of the favorable regions with operating conditions of the thermal reduction step (Reaction (2)) (a) Temperature.
2 First-principles predictions of descriptors
In this section, first-principles calculations were applied to the predictions of the descriptors (ΔSsolid, ΔHsolid) so that new redox materials can be added to the thermodynamic assessment map of Fig. 1.
In the first step, fast and reasonable predictions of (ΔSsolid, ΔHsolid) were demonstrated. ΔSsolid in Eq. (8) includes both vibrational (ΔSvib) and configurational (ΔSconf) entropic contributions[25].
Vibrational entropic contributions originate from the formations of defects, including oxygen vacancies as well as polarons[28,30], during the partial reduction step (Reaction (4)).
$2\text{Ce}_{\text{Ce}}^{\times }\text{+O}_{\text{O}}^{\times }\to \text{V}_{\text{O}}^{\centerdot \centerdot }+2\text{C}{{\text{{e}'}}_{\text{Ce}}}+\frac{1}{2}{{\text{O}}_{2}}$ Reaction (4)
where $\text{Ce}_{\text{Ce}}^{\times }$ and $\text{O}_{\text{O}}^{\times }$ represent Ce and O ions on their respective sites, $\text{V}_{\text{O}}^{\centerdot \centerdot }$ is a doubly ionized oxygen vacancy, and $\text{C}{{\text{{e}'}}_{\text{Ce}}}$ is an electron localized on a Ce ion, i.e. polaron. Therefore, ΔSvib is expressed in Eq. (9).
Where Sbulk, ${{S}_{\text{V}_{\text{O}}^{\centerdot \centerdot }}}$ and ${{S}_{\text{C}{{{\text{{e}'}}}_{\text{Ce}}}}}$ are the entropies of a bulk CeO2 supercell (Fig. 3(a)), a CeO2 supercell with a single oxygen-vacancy defect (Fig. 3(b)) and a CeO2 supercell with a single polaron defect (Fig. 3(c)), taking pure CeO2 as an example. More details are available in Supporting Materials on the preparations of the supercells and on the calculations of Sbulk, ${{S}_{\text{V}_{\text{O}}^{\centerdot \centerdot }}}$, ${{S}_{\text{C}{{{\text{{e}'}}}_{\text{Ce}}}}}$ and corresponding ΔSvib. Configurational entropic contributions are from the random distributions of ionic (oxygen vacancies) and electronic (polarons) defects, so ΔSconf was calculated using the ideal solution model (Methods in Supporting Materials). ΔHsolid was approximated as the oxygen-vacancy formation energy[11,21].
![]()
Figure 3.Supercells for DFT+
ΔSvib, ΔSconf and ΔHsolid of the CeO2/CeO2-δ and Ce0.9Sm0.1O1.95/ Ce0.9Sm0.1O1.95-δ redox pairs (δ=0.03) were calculated before the comparison of these calculation results with experimentally measured ΔSsolid and ΔHsolid[28-29,31]. Table 1 shows that this applied theoretical method can predict ΔSsolid and ΔHsolid of pure and Sm-doped ceria pairs, and expectedly other redox materials with reasonable accuracy. Particularly for ΔSsolid of the CeO2/CeO2-δ redox pair (δ= 0.03), it was demonstrated that the relative differences between calculated and measured values are below 12%, although it is found in references[20, 23] that this method might over-estimate ΔSsolid. Regarding ΔHsolid of pure and Sm-doped ceria pairs (δ=0.03), the relative differences are also below 12%, demonstrating sufficient accuracy of this method which is expected to be more efficient enabled by the advancement and power of high-throughput computational tools[15,16].
| Oxide | ΔVrel, vacancy/ nm3 | ΔVrel, polaron/nm3 |
|---|---|---|
| CeO2-δ | -0.0170 | 0.0139 |
| Ce0.9Sm0.1O1.95-δ | -0.0170 | 0.0144 |
Table 1.
Relaxation volumes due to formations of an oxygen vacancy and a polaron for CeO2-
Then physical insights were given into these calculation results to further demonstrate the reliability of the applied theoretical method. Fig. 4 shows the variations of $\Delta {{S}_{\text{V}_{\text{O}}^{\centerdot \centerdot }}}$, $2\Delta {{S}_{\text{C}{{{\text{{e}'}}}_{\text{Ce}}}}}$, ΔSvib and ΔSsolid with temperatures. Since the results for pure and Sm-doped ceria pairs follow the same trend, the CeO2/CeO2-δ redox pair is taken as an example. It is observed that $\Delta {{S}_{\text{V}_{\text{O}}^{\centerdot \centerdot }}}$ is negative, which can be intuitively understood from less atoms and resultantly lower vibrational degrees of freedom of the reduced oxides. This can also be quantitatively explained by a negative relaxation volume, ΔVrel, due to the formation of an oxygen-vacancy defect, from Eq. (S2) in Supporting Materials. The surrounding O ions relax towards the vacancy for a distance longer than the Ce ions relax away, which renders a relaxation volume of -0.0170 nm3 (Table S1). On the contrary, $2\Delta {{S}_{\text{C}{{{\text{{e}'}}}_{\text{Ce}}}}}$ is positive, which is resultant from a positive relaxation volume (ΔVrel=0.0139 nm3). These results indicate that the change of volume, due to the formation of defects, dominates vibrational entropic contribution. The calculated ΔSvib is 13.5 J∙(0.5 mol O2)-1∙K-1 at 1000 K which agrees well with the experimental data[28], and the calculated ΔSconf is 80.5 J∙(0.5 mol O2)-1∙K-1. For the Ce0.9Sm0.1O1.95/Ce0.9Sm0.1O1.95-δ redox pair, the fact that ΔSconf (70.3 J∙(0.5 mol O2)-1∙K-1) is smaller than that of pure ceria redox pair is reasonable because doping by an atom with a lower valence renders a reduction of possible microscopic states. ΔSsolid are 94.0 and 79.4 J∙(0.5 mol O2)-1∙K-1 for the CeO2/CeO2-δ and the Ce0.9Sm0.1O1.95/Ce0.9Sm0.1O1.95-δ redox pairs (δ=0.03). These values are comparable to 0.5SO2(121.8 J∙(0.5 mol O2)-1∙K-1) at 1000 K. These high positive ΔSsolid, attributed to the configurational and polaron defective entropic contributions, explain the excellent thermochemical performance of pure and doped ceria reported in literature [5-8].
![]()
Figure 4.Variations of $\Delta {{S}_{\text{V}_{\text{O}}^{\centerdot \centerdot }}}$, $2\Delta {{S}_{\text{C}{{{\text{{e}'}}}_{\text{Ce}}}}}$, Δ
3 Discussion
Based on the results of this work, a viable screening approach of materials for solar-driven CO2 splitting using two-step thermochemical cycles was proposed as shown in Fig. 5.
![]()
Figure 5.Illustration of a viable screening approach
The first step is to calculate the descriptors, the combination of ΔSsolid and ΔHsolid defined in Eq. (3) by the developed DFT+U based first-principles approach. As described in Introduction, despite the recognition of the entropic effect, energy descriptors (enthalpy of formation or energy of oxygen-vacancy formation) were usually used for the screening of material candidates in literature [11-12, 22]. Therefore, (ΔSsolid, ΔHsolid) represents a more general descriptor.
The second step is to screen material candidates by the derived criteria defined in Eq. (7) and find out qualified redox materials. As described in Section 1, these criteria were derived by comprehensively considering non-stoichiometric reaction mechanism, theoretical efficiency and practical operating conditions. It has to be pointed out that materials with (ΔSsolid, ΔHsolid) near the boundary of the favorable region (Fig. 1) should also be screened as qualified redox materials considering the accuracy of DFT calculations. In addition, Eq. (6) varies with target solar-to-chemical and thermal-to-chemical efficiencies as well as achievable solar-to-thermal efficiency. For example, ΔHsolid is ≤ 600 kJ∙(0.5 mol O2)-1 in Eq. (6) with the target solar-to-chemical efficiency reseted from 20% to 15%. Resultantly, the solid square scatter is inside the favorable region.
The third step is to perform reactor testing using qualified redox material. Thermodynamics of redox material candidates represents an initial screening criterion[23,32], which was demonstrated in this work. Reaction kinetics of redox material candidates, which is characterized in reactor testing, is the important subsequent criterion[33,34], although reaction kinetics is out of the scope of this work.
4 Conclusions
In summary, this work presented a rationale for thermodynamic and first-principles assessments of redox materials for solar-driven CO2 splitting using two-step thermochemical cycles. The combination of solid-state change of entropy (ΔSsolid) and enthalpy of formation (ΔHsolid) was used as the descriptor. Comprehensive criteria based on it were derived to obtain the (ΔSsolid, ΔHsolid) map. It was found that a triangular region in this map identified qualified material candidates featuring large positive ΔSsolid and small enough ΔHsolid. Furthermore, a DFT+U based first-principles approach was developed to fast and reasonably predict ΔSsolid and ΔHsolid for redox materials without available thermochemical data. This rationale was exemplified for pure and samaria-doped ceria.
Acknowledgments
The authors would like to acknowledge Dr. Steffen Grieshammer for helpful discussions on DFT calculations and Prof. Sheng Chen for VASP access.
Supporting materials
Supporting materials related to this article can be found at
Supporting materials:
FENG Qingying, LIU Dong, ZHANG Ying, FENG Hao, LI Qiang
(School of Energy and Power Engineering, Nanjing University of Science and Technology, Nanjing 210094, China)
Methods
where, N is the number of degrees of freedom, kB is the Boltzmann constant, ħ is the Planck constant, T is the temperature. The normalized phonon density of states, g(ω), at each phonon frequency, ω, was calculated using the open source package of Phonopy[S8]. Phonon calculations were performed for constant volume conditions (V=V0) where the stress is not relaxed due to defect formation, while entropies for constant pressure conditions (p=p0) in Eq. (9) were more concerned experimentally. They were obtained by Eq. (S2)[S9].
where αV is the volumetric thermal expansion coefficient, KT is the isothermal bulk modulus and ΔVrel is the relaxation volume, i.e., the difference between the volume of a defective supercell (oxygen-vacancy or polaron) and that of a bulk supercell. The free energy (E) of 11 unit cells with volumes (V) ranging from 0.03656 nm3 to 0.04656 nm3 was calculated for each temperature. These E-V curves for each temperature were fitted using the Birch-Murnaghan equation(Eq. (S3))[S9] to obtain KT and equilibrium volumes (V0) for each temperature.
Then, αV was derived from equilibrium volumes as Eq. (S4).
Calculated αV, KT and ΔVrel are available in Fig. S2 and Table S1.
Supporting figures and tables
![]()
Figure S1.Phonon dispersion of a 10% samaria-doped ceria supercell comprising an oxygen vacancy
![]()
Figure S2.Calculated variations of (a)
References:
[1]KRESSE G, FURTHMULLER J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B, 1996, 54: 11169-11186.
[2]PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple. Phys. Rev. Lett., 1997, 78: 18-28.
[3]BLOCHL P E. Projector augmented-wave method. Phys. Rev. B, 1994, 50: 17953-17979.
[4]DUDARV S L, BOTTON G A, SAVRASOV S Y, et al. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B, 1998, 57: 1505-1509.
[5]KOETTGEN J, GRIESHAMMER S, HEIN P, et al. Understanding the ionic conductivity maximum in doped ceria: trapping and blocking. Phys. Chem. Chem. Phys., 2018, 20: 14291-14321.
[6]ZACHERLE T, SCHRIEVER A, DE SOUZA R A, et al. Ab initio analysis of the defect structure of ceria. Phys. Rev. B, 2013, 87: 134104.
[7]GERWARD L, STAUN OLSEN J, PETIT L, et al. Bulk modulus of CeO2 and PrO2-an experimental and theoretical study. J. Alloy Compd., 2005, 400: 56-61.
[8]GINZE X, LEE C. Dynamical matrices, born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory. Phys. Rev. B, 1997, 55: 10355-10368.
[9]GRIESHAMMER S, ZACHERLE T, MARTIN M. Entropies of defect formation in ceria from first principles. Phys. Chem. Chem. Phys., 2013, 15: 15935-15942.
[10]KOBAYASHI T, WANG S, DOKIYA M, et al. Oxygen nonstoichiometry of Ce1-ySmyO2-0.5y-x (y=0.1, 0.2). Solid State Ionics, 1999, 126: 349-357.
References
[17] S GAUTAM G, A CARTER E. Evaluating transition-metal oxides within DFT-SCAN and SCAN+U frameworks for solar thermochemical applications. Phys. Rev. Mater., 2, 095401(2018).
[19] R SHAH P, T KIM, G ZHOU et al. Evidence for entropy effects in the reduction of ceria-zirconia solutions. Chem. Mater., 18, 5363-5369(2006).
[23] B GOPAL C, A VAN DE WALLE.
[26] W CHASE M. NIST-JANAF Thermochemical Tables, 4th Edition. New York: American Institute of Physics, 647-1745(1998).


Set citation alerts for the article
Please enter your email address

© Copyright 2018-2021 | Chinese Laser Press. All Rights Reserved 沪ICP备15018463号-20