
Bingying Zhao, Jerome Mertz, "Resolution enhancement with deblurring by pixel reassignment," Adv. Photon. 5, 066004 (2023)
- Advanced Photonics
- Vol. 5, Issue 6, 066004 (2023)
Abstract
1 Introduction
The spatial resolution of conventional fluorescence microscopy is limited to about half the emission wavelength because of diffraction.1 This limit can be surpassed using a variety of superresolution approaches. For example, techniques such as STED,2
In recent years, it has been noted that the sparsity constraint can be partially alleviated by pre-sharpening the raw images. Example algorithms are SRRF30,31 and MSSR,32 which are freely available and easy to use. In contrast to DeconSTORM, these algorithms make only minimal assumptions about the emission PSF (radiality in the case of SRRF; convexity in the case of MSSR), and their application can substantially reduce the number of raw images required for SMLM. Indeed, when applied to denser images, only few images or even a single raw image can produce results quite comparable to much more time-consuming superresolution approaches. However, these algorithms are not without drawbacks. SRRF and MSSR are both inherently highly nonlinear, meaning that additional steps are required to enforce a linear relation between sample and image brightness.30
We present an alternative image-sharpening approach that is similar to SRRF and MSSR, but has the advantage of inherently preserving image intensities and being more generally applicable. Like SRRF and MSSR, our approach can be applied to a wide variety of fluorescence microscopes, where we make only minimal assumptions about the emission PSF, namely, that the PSF centroid is located at its peak. Also, like SRRF and MSSR, our approach can be applied to a sequence of raw images, allowing a temporal analysis of blinking or fluctuation statistics, or it can be applied to only a few or even a single image. Our approach is based on postprocessing by pixel reassignment, producing a deblurring effect similar to deconvolution but without the drawbacks associated with conventional deconvolution algorithms. We describe the basic principle of deblurring by pixel reassignment (DPR) and compare its performance to SRRF and MSSR both experimentally and with simulated data. Our DPR algorithm is made available as a MATLAB function.
Sign up for Advanced Photonics TOC. Get the latest issue of Advanced Photonics delivered right to you!Sign up now
2 Principle of DPR
Fundamental to any linear imaging technique is the concept of a PSF: point sources in a sample produce light distributions at the imaging plane that are blurred by a convolution operation with the PSF. Because the width of the PSF is finite, so too is the image resolution. In principle, if the PSF is known exactly, the blurring caused by convolution can be undone numerically by deconvolution; however, in practice, such deblurring is hampered by fundamental limitations. For one, the Fourier transform of the PSF (or optical transfer function—OTF) provides a spatial frequency support inherently limited by the finite size of the microscope pupil, meaning that spatial frequencies beyond this diffraction limit are identically zero and cannot be recovered by deconvolution, even in principle (unless aided by assumptions,33
The purpose of DPR is to perform PSF sharpening similar to deconvolution, but in a manner less prone to noise-induced artifacts and without the requirement of a full model for the PSF. Unlike Wiener deconvolution, which is performed in Fourier space using a division operation, DPR operates entirely in real space with no division operation that can egregiously amplify noise. Unlike RL deconvolution, DPR is noniterative and can be performed in a single pass, without the need for an arbitrary iteration-termination criterion. DPR relies solely on pixel reassignment. As such, no negativities are possible in the final image reconstruction, as is often encountered, for example, with Wiener deconvolution or image sharpening with a Laplacian filter.39 Moreover, intensity levels are rigorously conserved, with no requirement of additional procedures to ensure local linearity, as needed, for example, with SRRF, MSSR, or even SOFI.40
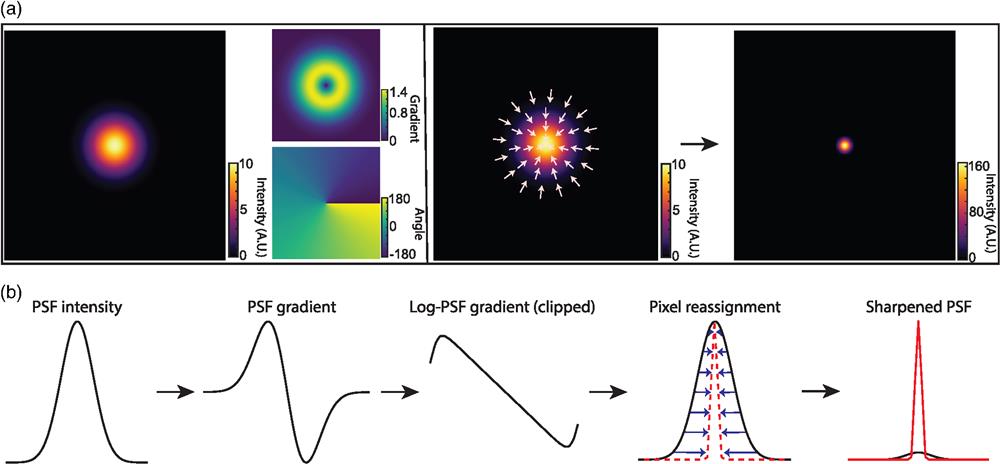
The basic principle of DPR is schematically shown in Fig. 1 and described in more detail in Sec. 5. In brief, raw fluorescence images are first preconditioned by (1) performing global background subtraction, (2) normalizing to the overall maximum value in the image, and (3) re-mapping by interpolation to a coordinate system of grid period given by roughly one-eighth of the full width at half-maximum (FWHM) of the PSF. The purpose of such preconditioning is to standardize the raw images prior to the application of DPR. The actual sharpening of the image is then performed by pixel reassignment, where intensities (pixel values) at each grid location (pixel) are reassigned to neighboring locations according to the direction and magnitude of the locally normalized image gradient (or, equivalently, the log-image gradient), scaled by a gain parameter. Because pixels are generally reassigned to off-grid locations, their pixel values are distributed to the nearest on-grid reassigned locations as weighted by their proximity (see Fig. S1 in the Supplementary Material). Finally, an assurance is included that pixels can be displaced no farther than 1.25 times the PSF FWHM width.
![]()
Figure 1.Principle of DPR. (a) From left to right: simulations of Gaussian PSF intensity and gradient maps (amplitude and direction), pixel reassignments, deblurred PSF image after application of DPR. (b) DPR workflow.
As a simple example, consider imaging a point source with a Gaussian PSF of root-mean-square (RMS) width
Conventionally, the resolution of a microscope is defined by its capacity to distinguish two point sources. More specifically, it is defined by the minimum separation distance required for two points to be resolved based on a predefined criterion, such as the Sparrow or Rayleigh criterion. We again consider the example of a Gaussian PSF, but now with two point sources. According to the Sparrow and Rayleigh criteria, the two points would have to be separated by
Similar resolution enhancement results are obtained when DPR is applied to two line objects. Here, we use raw data acquired by an Airyscan microscope obtained from Refs. 32 and 41 [Fig. 2(b)]. In the raw image, lines separated by 150 nm cannot be resolved, whereas after the application of DPR with gains 1 and 2, they can be resolved at separations of 90 and 30 nm, respectively. The intensity profiles across the full set of line pairs for raw, DPR gain 1, and DPR gain 2 images are shown in Fig. S4(a) in the Supplementary Material. DPR images of the same sample acquired by conventional confocal microscopy32,41 are shown in Fig. S4(b) in the Supplementary Material. In this case, lines separated by 210 nm cannot be resolved in the raw data set, whereas after application of DPR with gains 1 and 2, they can be resolved at separations of 120 and 90 nm, respectively.
![]()
Figure 2.DPR resolution enhancement. (a) Simulation of DPR applied to two point objects separated by
To gain an appreciation of the effect of noise on DPR, we again simulated images of two point objects and two line objects, this time separated by 160 nm and imaged with a Gaussian PSF of RMS 84.93 nm. To these images we added shot noise (Poissonian) and additive camera readout noise (Gaussian) of different strengths, leading to SNR values of 5.0, 7.7, 14.1, and 20.3 (see Fig. S5 in the Supplementary Material). DPR gain 1 was applied to a stack of images, each with a different noise realization. The resulting resolution-enhanced images were then averaged over different numbers of frames (10, 20, and 40). Manifestly, the final image quality improves with increasing SNR and/or increasing numbers of frames averaged, as expected. Accordingly, the error in the measured separation between the two point objects and the two line objects as inferred by the separation between their peaks in the images also decreases [see Fig. S5(c) in the Supplementary Material]. The images of line objects were less sensitive to noise, as evidenced by the relatively stable separation errors across various SNRs, but they exhibited somewhat higher separation error compared to the images of the two point objects. These results are qualitative only. Nevertheless, they provide a rough indication of the increase in enhancement fidelity with SNR.
3 Results
3.1 DPR Applied to Single-Molecule Localization Imaging
To demonstrate the resolution enhancement capacity of DPR with experimental data, we applied it to SMLM images. We used raw images made publicly available through the SMLM Challenge 2016,42 as these provide a convenient standardization benchmark. The experimental data consisted of a 4000-frame sequence of STORM images of microtubules labeled with Alexa567.
We applied DPR separately to each frame. Similar to SRRF and MSSR, we included in our DPR algorithm the possibility of temporal analysis of DPR-enhanced images. Here, the temporal analysis is simple and consists either of averaging in time the DPR-enhanced images or calculating their variance in time (as is done, for example, with SOFI imaging of order 2). The results are shown in Figs. 3(a) and 3(b). As expected, DPR gain 2 leads to greater resolution enhancement than gain 1. Moreover, as expected, the temporal variance analysis leads to enhanced image contrast, since it preferentially preserves fluctuating signals while removing nonfluctuating backgrounds. However, it should be noted that temporal variance analysis no longer preserves a linearity between sample and image strengths, as opposed to temporal averaging.
![]()
Figure 3.SMLM Challenge 2016. (a) DPR applied to each frame in raw image stack, followed by temporal mean or variance. (i) Raw image stack, (ii) mean of raw images,, (iii) DPR gain 1 followed by mean, (iv) DPR gain 2 followed by mean, (v) DPR gain 1 followed by variance, and (vi) DPR gain 2 followed by variance. Scale bar, 650 nm. (b) Expanded regions of interest (ROIs) indicated by green square in (a). Bottom left, intensity distribution along red line in ROIs. Bottom right, intensity distribution along green line in ROIs. Scale bar, 200 nm. (c) Image mean followed by DPR. (vii) gain 1, (viii) gain 2. Scale bar, 500 nm. (d) Expanded ROIs indicated by yellow squares in (c) and (ii), (iii), and (iv) in (a). Right, intensity distribution along cyan line in ROIs. Scale bar, 150 nm. PSF FWHM, 2.7 pixels. Local-minimum filter radius, 5 pixels.
Interestingly, when a temporal average was applied to the raw images prior to the application of DPR [i.e., when the order of DPR and averaging was reversed—Figs. 3(c) and 3(d)], DPR continued to provide resolution enhancement, but not as effectively as when DPR was applied separately to each raw frame. The reason for this is clear. DPR relies on the presence of spatial structure in the image, which is largely washed out by averaging. In other words, similar to SRRF and MSSR, DPR is most effective when imaging sparse samples, as indeed is a requirement for SMLM.
3.2 DPR Maintains Imaging Fidelity
DPR reassigns pixels according to their gradients. If the gradients are zero, the pixels remain in their initial position. That is, when imaging structures larger than the PSF that present gradients only around their edges but not within their interior, DPR sharpens only the edges while leaving the structure interiors unchanged. This differs, for example, from SRRF or MSSR, which erode or hollow out the interiors erroneously. DPR can thus be applied to more general imaging scenarios where samples contain both small and large structures. This is apparent, for example, when imaging a Siemens star target, as shown in Fig. S6 in the Supplementary Material, where neither SRRF nor MSSR accurately represents the widening of the star spokes. For example, when we applied NanoJ-SQUIRREL43 to the DPR-enhanced, MSSR-enhanced, and SRRF-enhanced Siemens star target images, we found resolution-scaled errors43 (RSEs) given by 53.5, 95.4, and 102.6, respectively; and resolution-scaled Pearson coefficients43 (RSPs) given by 0.92, 0.54, and 0.75, respectively. This improved fidelity is apparent, also, in other imaging scenarios. In Fig. 4, we show results obtained from the image of Alexa488-labeled bovine pulmonary artery endothelial (BPAE) cells (ThermoFisher, FluoCells) acquired with a conventional laser scanning confocal microscope (Olympus FLUOVIEW FV3000; objective,
![]()
Figure 4.Comparison of DPR, MSSR, and SRRF performances. (a) Images of BPAE cells acquired by a confocal microscope and after DPR gain 2, first order MSSR, and SRRF. DPR parameters: PSF FWHM, 4 pixels; local-minimum filter radius, 12 pixels; DPR gain, 2. MSSR: PSF FWHM, 4 pixels; magnification, 2; order, 1. SRRF: ring radius, 0.5, magnification, 2;, axes, 6. Scale bar,
A difficulty when evaluating image fidelity is the need for a ground truth as a reference. To serve as a surrogate ground truth, we obtained images of BPAE cells with a state-of-the-art Nikon CS-WU1 spinning disk microscope equipped with a superresolution module (SoRa) based on optical pixel reassignment44 (objective,
![]()
Figure 5.Comparison of DPR, SRRF, and MSSR with optical pixel reassignment and deconvolution. (a) Left, BPAE cells imaged using optical pixel reassignment and deconvolution with Nikon confocal microscope without (top) and with SoRa superresolution enhanced by RL deconvolution (20 iterations). Right, confocal images deblurred by RL deconvolution (20 iterations), DPR (gains 1 and 2), SRRF, and MSSR. DPR parameters: PSF FWHM, 2 pixels; local-minimum filter radius, 40 pixels. MSSR parameters: PSF FWHM, 2 pixels; magnification, 4; order, 1. SRRF: ring radius, 0.5; magnification, 4; axes, 6. Scale bar,
3.3 DPR Applied to Engineered Cardiac Tissue Imaging
To demonstrate the ability of DPR to enhance image information, we performed structural imaging of engineered cardiac micro-bundles derived from human-induced pluripotent stem cells (hiPSCs), which have recently gained interest as model systems to study human cardiomyocytes (CMs).45,46 We first imaged a monolayer of green fluorescent protein (GFP)-labeled hiPSC-CMs with a confocal microscope of sufficient resolution to reveal the
![]()
Figure 6.Engineered cardiac tissue imaging. (a) DPR gain 1 applied to simulated ground-truth wide-field images of monolayer hiPSC-CMs derived from experimental images acquired by a confocal microscope. Left, simulated ground truth. Middle, simulated wide-field intensity image without (top) and with (bottom) application of DPR, and corresponding error maps. Right, intensity profile along sarcomere chain indicated by the cyan rectangle. PSF FWHM, 4 pixels; local-minimum filter radius, 7 pixels. (b) DPR gain 1 applied to experimental low-resolution images of hiPSC-CMTs. Left, confocal image. Middle, DPR-enhanced image. Right, intensity profile of sarcomere chain indicated by the red rectangle. PSF FWHM, 4 pixels; local-minimum filter radius, 7 pixels. (c) DPR gain 1 applied to experimental high-resolution images of hiPSC-CMT. Left, confocal image. Middle, DPR-enhanced image. Right, intensity profile of the sarcomere chain indicated by the yellow rectangle. PSF FWHM, 2 pixels; local-minimum filter radius, 4 pixels. Scale bar,
We also performed imaging of hiPSC cardiomyocyte tissue organoids (hiPSC-CMTs). Such imaging is more challenging because of the increased thickness of the organoids (about
3.4 DPR Applied to Volumetric Zebrafish Imaging
In recent years, there has been a push to develop microscopes capable of imaging populations of cells within extended volumes at high spatiotemporal resolution. One such microscope is based on confocal imaging with multiple axially distributed pinholes, enabling simultaneous multiplane imaging.48,49 However, in its most simple implementation, multi-z confocal microscopy is based on low NA illumination and provides only limited spatial resolution, roughly
![]()
Figure 7.Multi-z confocal zebrafish imaging. (a)
4 Discussion
The purpose of DPR is to help counteract the blurring induced by the PSF of a fluorescence microscope. The underlying assumption of DPR is that fluorescent sources are located by their associated PSF centroids, which are found by hill climbing directed by local intensity gradients.51,52 When applied to individual fluorescence images, DPR helps distinguish nearby sources, even when these are separated by distances smaller than the Sparrow or Rayleigh limit. In other words, DPR can provide resolution enhancement even in densely labeled samples. Such resolution enhancement is akin to image sharpening, with the advantage that DPR is performed in real space rather than Fourier space, and that local intensities are inherently preserved and negativities are impossible (Table S1 in the Supplementary Material).
To define what is meant by the term local here, we can directly compare the intensities of raw and DPR-enhanced images. When both images are spatially filtered by average blurring, the differences between their intensities become increasingly small with increasing kernel size of the average filter (see Fig. S10 in the Supplementary Material). Indeed, the relative differences, as characterized by the difference standard deviations, drop to
Of course, no deblurring strategy is immune to noise, and the same is true for DPR. However, DPR presents the advantage that noise cannot be amplified as it can be, for example, with Wiener or RL deconvolution, both of which require some form of noise regularization (in the case of RL, iteration termination is equivalent to regularization). DPR requires neither regularization nor even an exact model of the PSF. As such, DPR resembles SRRF and MSSR, but with the advantage of simpler implementation and more general applicability to samples possessing extended features.
Finally, our DPR algorithm is made available here as a MATLAB function compatible with either Windows or MacOS. An example time to process a stack of
Because of its ease of use, speed, and versatility, we believe DPR can be of general utility to the bio-imaging community.
5 Methods
5.1 DPR Algorithm
Figure S9 in the Supplementary Material illustrates the overall workflow of DPR, and Algorithm S1 in the Supplementary Material provides more details in the form of a pseudo-code. To begin, we subtract the global minimum from each raw image to remove uniform background and camera offset (if any). These background-subtracted images
Next, we must establish a vector map to guide the pixel reassignment process. This is done in steps. First, we perform local equalization of
The reassignment vector map for DPR is obtained by first calculating the gradients of
Pixel reassignment consists of numerically displacing the intensity values in the input image from their initial grid position to new reassigned positions according to their associated pixel reassignment vector. In general, as shown in Fig. S1 in the Supplementary Material, the new reassigned positions are off-grid. The pixel values are then partitioned to the nearest four on-grid locations as weighted by their proximity to these locations, as described in more detail in Algorithm S1 in the Supplementary Material.
Reassignment is performed pixel-by-pixel across the entire input image, leading to a final output DPR image. In the event that a time sequence of the image is processed, the output DPR sequence can be temporally analyzed (for example, by calculating the temporal average or variance) if desired. Note that an input parameter for our DPR algorithm is the estimated PSF size. When using the Olympus FV3000 microscope, we obtained this from the manufacturer’s software. When using our home-built confocal microscopes, we measured this with 200 nm fluorescent beads (Phosphorex). When using the SoRa microscope, we used the estimated PSF for confocal microscopy. The PSF size need not be exact and may be estimated from the conventional Rayleigh resolution limit given as55
5.2 Simulated Data
Simulated wide-field images of two point objects and two line objects separated by 160 nm were used to evaluate the separation accuracy of DPR using Gaussian PSF of standard deviation 84.93 nm. The images were rendered on a 40 nm grid. Poisson noise and Gaussian noise were added to simulate different SNRs, with SNR being calculated as
Simulated wide-field images of the sarcomere ground truth were produced based on a Gaussian PSF model of standard deviation
5.3 Engineered Heart Tissue Preparation
hiPSCs from the PGP1 parent line (derived from PGP1 donor from the Personal Genome Project) with an endogenous GFP tag on the sarcomere gene TTN56 were maintained in mTeSR1 (StemCell) on Matrigel (Fisher) mixed 1:100 in DMEM/F-12 (Fisher) and split using accutase (Fisher) at 60% to 90% confluence. hiPSCs were differentiated into monolayer hiPSC-CMs by the Wnt signaling pathway.57 Once cells were beating, hiPSC-CMs were purified using RPMI no-glucose media (Fisher) with 4 mmol/L sodium DL lactate solution (Sigma) for 2 to 5 days. Following selection, the cells were replated and maintained in RPMI with 1:50 B-27 supplement (Fisher) on
Three-dimensional (3D) hiPSC-CMTs devices with tissue wells, each containing two cylindrical micropillars with spherical caps, were cast in PDMS from a 3D-printed mold (Protolabs).58 A total of 60,000 cells per CMT, 90% hiPSC-CMs, and 10% normal human ventricular cardiac fibroblasts were mixed in
5.4 Zebrafish Preparation
All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Boston University, and practices were consistent with the Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act. For the in vivo structural imaging of zebrafish, transgenic zebrafish embryos (isl2b:Gal4 UAS:Dendra) expressing GFP were maintained in filtered water from an aquarium at 28.5°C on a 14 to 10 h light–dark cycle. Zebrafish larvae at 9 days postfertilization (dpf) were used for imaging. The larvae were embedded in 5% low-melting-point agarose (Sigma) in a 55 mm petri dish. After agarose solidification, the petri dish was filled with filtered water from the aquarium.
5.5 hiPSC-CMTs Imaging
The hiPSC-CMTs were imaged with a custom confocal microscope, essentially identical to that described in Ref. 48, but with adjustable illumination NA (0.2 NA with
5.6 DPR, MSSR, and SRRF Parameters
The parameters used for DPR, MSSR, and SRRF for our results can be found in Table S2 in the Supplementary Material.
5.7 Error Map Calculation
Error map calculation is realized by a custom script written in MATLAB R2021b. Pixelated differences between the images and the ground truth are directly measured by subtraction and saved as an error map.
5.8 SSIM Calculation
The SSIM calculation is realized by the SSIM function in MATLAB R2021b. Exponents for luminance, contrast, and structural terms are set as [1, 1, 1] (default values). Standard deviation of the isotropic Gaussian function is set as 1.5 (default value).
Bingying Zhao is currently a PhD student in the Department of Electrical and Computer Engineering at Boston University. She received her BS degree in optical information science and technology from Sun Yat-sen University and her MS degree in optics and photonics from Karlsruhe Institute of Technology in 2016 and 2019, respectively. Her current research focuses on biomedical imaging.
Jerome Mertz is a professor of biomedical engineering and director of the Biomicroscopy Laboratory at Boston University. Prior to joining Boston University, he was a CNRS researcher at the École Supérieure de Physique et de Chimie Industrielles in Paris. His research interest includes the development and applications of novel optical microscopy techniques for biological imaging. He is a fellow of the Optical Society and the American Institute for Medical and Biological Engineering.
References
[1] J. Mertz. Introduction to Optical Microscopy(2019).
[4] S. W. Hell. Toward fluorescence nanoscopy. Nat. Biotechnol., 21, 1347-1355(2003).
[14] C. B. Müller, J. Enderlein. Image scanning microscopy. Phys. Rev. Lett., 104, 198101(2010).
[15] S. Roth et al. Optical photon reassignment microscopy (OPRA). Opt. Nanosc., 2, 5(2013).
[25] F. Chen, P. W. Tillberg, E. S. Boyden. Expansion microscopy. Science, 347, 543-548(2015).
[26] I. Cho, J. Y. Seo, J. Chang. Expansion microscopy. J. Microsc., 271, 123-128(2018).
[36] N. Wiener. Extrapolation, Interpolation, and Smoothing of Stationary Time Series: With Engineering Applications(1949).
[41] R. D’Antuono. Airyscan and confocal line pattern(2022).
[53] I. J. Schoenberg. Cardinal Spline Interpolation(1973).
[55] J. Pawley. Handbook of Biological Confocal Microscopy(2006).
[58] J. Javor et al. Controlled strain of cardiac microtissue via magnetic actuation, 452-455(2020).


Set citation alerts for the article
Please enter your email address

© Copyright 2018-2021 | Chinese Laser Press. All Rights Reserved 沪ICP备15018463号-20