Abstract
Synthesis pressure and structural stability are two crucial factors for highly energetic materials, and recent investigations have indicated that cerium is an efficient catalyst for N2 reduction reactions. Here, we systematically explore Ce–N compounds through first-principles calculations, demonstrating that the cerium atom can weaken the strength of the N≡N bond and that a rich variety of cerium polynitrides can be formed under moderate pressure. Significantly, -CeN6 possesses the lowest synthesis pressure of 32 GPa among layered metal polynitrides owing to the strong ligand effect of cerium. The layered structure of -CeN6 proposed here consists of novel N14 ring. To clarify the formation mechanism of -CeN6, the reaction path Ce + 3N2 → trans-CeN6 → -CeN6 is proposed. In addition, -CeN6 possesses high hardness (20.73 GPa) and can be quenched to ambient conditions. Charge transfer between cerium atoms and N14 rings plays a crucial role in structural stability. Furthermore, the volumetric energy density (11.20 kJ/cm3) of -CeN6 is much larger than that of TNT (7.05 kJ/cm3), and its detonation pressure (128.95 GPa) and detonation velocity (13.60 km/s) are respectively about seven times and twice those of TNT, and it is therefore a promising high-energy-density material.I. INTRODUCTION
Polymeric nitrogen materials are environmentally friendly high-energy-density materials (HEDMs). Their high energy density comes from the huge energy difference between the N≡N triple bond (954 kJ/mol) on the one hand and the N=N double and N–N single bonds (418 and 160 kJ/mol, respectively) on the other. To date, a wide variety of novel polynitrogen structures have been reported in theoretical studies, such as the three-dimensional (3D) network structures (cg-N, Pnnm, Cccm, and CW),1–3 two-dimensional (2D) layer structures (A7, ZS, LB, LP, HLP, PP, and BP phases),4–9 one-dimensional (1D) chain structures (ch-N and Cmcm),10 and a zero-dimensional (0D) cage structure (N10).11 High temperature and high pressure are two important factors in obtaining polynitrogen materials. However, the ultrahigh pressure (>100 GPa) and temperature (>2000 K) in experiment required for the synthesis of cg-N, LP-N, HLP-N, and BP-N limit their development.12–15 An additional challenge is posed in quenching the high-pressure phases to ambient conditions, owing to their intrinsic metastable properties. Therefore, reducing the synthesis pressure and enhancing the structural stability of polynitrogen materials are crucial for their practical application. Nitrogen-rich compounds have been demonstrated to be promising candidates to achieve these goals.
In experiments, LiN5, K2N6, MgN4, Mg2N4, CsN5, α-ZnN4, β-ZnN4, and tr-BeN4 have been synthesized at pressures of 45, 45, 50, 50, 60, 63.5, 81.7, and 85 GPa, respectively.16–21 Moreover, LiN5, Mg2N4, and tr-BeN4 can be quenched down to ambient conditions. Clearly, compared with polynitrogen structures, nitrogen-rich compounds have milder synthesis conditions and higher stability. Theoretically, they exhibit a wide variety of nitrogen configurations, ranging from N2 dumbbells,22,23 azide roots,24–26 N rings (N4,27,28 N5,29–34 and N635–37), 1D N chains,38–43 2D N layers,44–49 to 3D network structures.50,51 Recently, the lanthanide polynitrides -GdN6 and -ErN6 have been proposed as HEDMs with excellent explosive performance.52,53 As a typical lanthanide element, cerium (Ce) is the most abundant rare earth element in the Earth’s crust and much cheaper than most rare earth metals, such as Gd and Er. Additionally, Ce atoms possess flexible electronic properties that allow them to reach high coordination numbers in their compounds. Thus, they exhibit excellent performance in the selective catalytic reduction of N2 to NH3.54–56 In light of these characteristics, we suggest that Ce may be an ideal candidate for inducing polynitrogen structures. However, up until now, only CeN has been reported in high-pressure studies,57–59 which has motivated us to perform a systematic high-pressure study on nitrogen-rich Ce–N compounds.
In this work, we systematically study Ce–N high-pressure compounds at pressures up to 100 GPa through first-principles swarm-intelligence structural searches. Seven stoichiometric ratios of CeNn (n = 0.5, 1, 2, 3, 4, 5, and 6) compounds are explored. Six new high-pressure phases are proposed, and their stability is verified using phonon dispersion curves, elastic constants, and molecular dynamic simulations. Interestingly, -CeN6 not only possesses the lowest synthesis pressure of 32 GPa among layered metal polynitrides, but also can be quenched to ambient conditions. The stability mechanism of -CeN6 is clarified by electronic structure and bonding analyses, and its mechanical properties are analyzed. Its infrared spectra (IR) and Raman spectra are calculated for experimental reference. Significantly, -CeN6 shows excellent volumetric energy density (up to 11.20 kJ/cm3), detonation pressure (128.95 GPa), and detonation velocity (13.60 km/s), opening up exciting avenues for the exploration of high-nitrogen-concentration lanthanide polynitrides via the metal ligand effect.
II. CALCULATION DETAILS
The structure searches were performed using the particle swarm optimization structure prediction method in the CALYPSO code.60 Seven stoichiometric ratios of CeNn (n = 0.5, 1, 2, 3, 4, 5, and 6) compounds were considered in the structural prediction at 0, 20, 50, and 100 GPa. The simulation cells contained 1, 2, and 4 formula units (f.u.), in which a total of 75 600 structures were produced and were ranked according to the calculated enthalpy. The previously reported -CeN and P4/nmm-CeN were successfully reproduced in our search,58 validating the effectiveness of the method. The structural relaxations and property calculation were performed using the Vienna Ab initio Simulation Package (VASP).61 The generalized gradient approximation (GGA) was used with the Perdew–Burke–Ernzerhof (PBE) exchange correlation functional.62 The GGA + U method (U = 6 eV) was used to correct the strong on-site Coulomb repulsion of Ce_4f states.63–66 The valence electrons of Ce and N atoms in the projector augmented wave (PAW) pseudopotentials were 4f15d16s2and 2s22p3, respectively.67 The Monkhorst–Pack k mesh spacing density and plane wave energy cutoff were set to 2π × 0.03 Å−1 and 520 eV, respectively. The accurate band structure was obtained using a hybrid functional (HSE06).68 The enthalpies of formation ΔHf of the CeNn compounds were calculated using the equation
The phonon dispersion curves were obtained using the PHONOPY code with density-functional perturbation theory.69 The energy density was calculated by considering the following dissociation path under ambient pressure: . The detonation velocity and detonation pressure were calculated from the Kamlet–Jacobs semiempirical equations70 and . The ab initio molecular dynamics (AIMD) simulations were performed in the isobaric–isothermic (NPT) ensemble with a total simulation time of 10 ps.71 The crystal orbital Hamilton population (COHP) was calculated using the LOBSTER package.72 For Raman and IR spectra, norm-conserving pseudopotentials were adopted, and these were calculated using the CASTEP module of the Material Studio package. Convergence criteria consisted of an energy change of <2 × 10−5 eV/atom and a maximum force of <0.05 eV/Å.73
III. RESULTS AND DISCUSSION
A. Phase diagram and structural stability
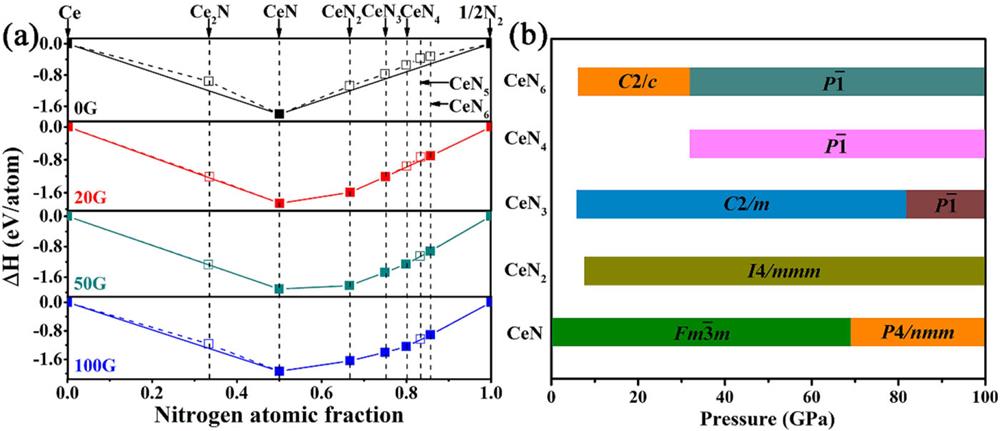
The convex hull of CeNn compounds is calculated for screening the thermodynamically stable phases of optimal structures in the prediction, and the results show that Ce can promote the formation of various polynitrides under high pressure. The stable phases of solid cerium ( and Cmcm phases) and solid nitrogen (, P42/mnm, P41212, and cg-N phases) are used as the references to calculate the formation enthalpy ΔHf, and this is plotted in Fig. 1(a), where the solid and blank squares correspond to the thermodynamically stable and unstable/metastable phases, respectively. The phase diagrams of thermodynamically stable high-pressure structures are obtained from the enthalpy difference analysis and are shown in Fig. 1(b) and Fig. S1 in the supplementary material. It can be seen that -CeN is stable from 0 to 69 GPa and then transforms to P4/nmm-CeN. This phase transition pressure is consistent with a previous experimental result (65 GPa).58I4/mmm-CeN2 remains stable at 8–100 GPa. C2/m-CeN3 is stable above 6 GPa, and then changes to -CeN3 at 82 GPa. As the nitrogen concentration increases, -CeN4 remains stable in the pressure range of 32–100 GPa. At 20 and 50 GPa, we predicted C2/c-CeN6 and -CeN6 phases, which are energetically favorable at 9–32 GPa and 32–100 GPa, respectively. Interestingly, as far as we know, the synthesis pressure (32 GPa) of -CeN6 is the lowest among the layered metal polynitrides.
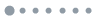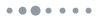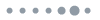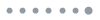
Figure 1.(a) Formation enthalpies ΔH of various CeNn (n = 0.5, 1, 2, 3, 4, 5, and 6) compounds under high pressure. The stable phases are connected by solid lines and unstable/metastable phases by dashed lines. (b) Pressure–composition phase diagram of the predicted Ce–N phases.
The crystal structures of CeNn compounds reveal that Ce has strong coordination ability under high pressure [Figs. 2 and S2 (supplementary material)], with each Ce atom coordinating with 10, 8, 10, 10, 10, and 12 N atoms in I4/mmm-CeN2, C2/m-CeN3, -CeN3, -CeN4, C2/c-CeN6, and -CeN6, respectively [Fig. S3 (supplementary material)]. The N-structure units in I4/mmm-CeN2 and C2/c-CeN6 are N2 dumbbells, and those in C2/m-CeN3 and -CeN3 are N2 dumbbells and N4 molecular chains. The polymeric structures of -CeN4 and -CeN6 are N8 molecular chains and layered structures with an N14 ring, respectively [Fig. S4 (supplementary material)]. Calculations of phonon dispersion curves show that the six new CeNn compounds described above are dynamically stable at predicted pressures owing to the absence of an imaginary frequency in the Brillouin zone [Fig. S5 (supplementary material)], and thus they are high-pressure stable phases. The structural parameters are listed in Table SI (supplementary material). Excluding C2/c-CeN6, the average N–N bond length in CeNn compounds is about 1.30–1.39 Å [Table SII (supplementary material)], which is much larger than that in nitrogen molecules (1.11 Å), suggesting that these represent a new type of nitrogen-rich materials in which the N–N bonds are single or intermediate between single and double in nature. We deduce that they may have high energy capacity properties.
Among these CeNn compounds, -CeN6 is particularly fascinating and worthy of further discussion because of its high nitrogen concentration, long N–N bonds, high coordination number, and low synthesis pressure. As shown in Fig. 2, the crystal structure of -CeN6 is layered, with an AαAα stacking form along the a-axis direction, where A and α represent the N plane and Ce plane, respectively. Interestingly, -CeN6 possesses unique N14 rings, the first to be reported up until now. At 32 GPa, the bond lengths of N1–N1, N1–N3, N3–N3, N3–N2, and N2–N2 are 1.40, 1.42, 1.47, 1.35, and 1.45 Å, respectively, indicating that the N3–N2 bond is intermediate in nature between a single bond (1.45 Å) and a double bond (1.25 Å), whereas all the other bonds are close to single bond in nature. The mechanical and dynamical stability of -CeN6 were verified at 32 GPa by calculating elastic constants and phonon dispersion curves [Fig. 2(c) and Table SIII (supplementary material)]. The phonon density of states (PHDOS) in Fig. 2(c) shows that the high-frequency vibrational modes come from N–N vibrations, while the low-frequency vibrational modes consist of Ce–N collective motions. Moreover, the thermal stability of -CeN6 was evaluated using AIMD simulations. As shown in Fig. 2(d), the total energy of -CeN6 fluctuates around the equilibrium position (−751.92 eV), and the structural skeleton remains intact at the end of the simulation, suggesting that -CeN6 is thermally stable under a pressure of 32 GPa and a temperature of 300 K.
Figure 2.(a) Crystal structure of -CeN6 at 32 GPa. (b) Nitrogen structural skeleton of -CeN6 at 32 GPa. The independent N atoms are marked as N1, N2, and N3 in the symmetric N14 rings. (c) Phonon dispersion curve and PHDOS of -CeN6 at 32 GPa. (d) Initial and terminal structures in AIMD simulations with total simulation time 10 ps and fluctuations of the total energy of -CeN6 at 32 GPa and 300 K.
B. Stability mechanism under high pressure
To pinpoint the stability mechanism of -CeN6, we carefully analyzed the electron localization function (ELF). As shown in Fig. 3(a), the N1 atoms in -CeN6 hybridize in sp2 states with two σ bonds and a lone electron pair. Both the N2 and N3 atoms are sp3 hybridized. The hybrid orbital of N2 atoms contains two σ bonds and two lone electron pairs, and that of the N3 atom contains three σ bonds and a lone electron pair. Additionally, the strong localization of electrons between N atoms indicates a strong N–N covalent bond interaction, and the lone-pair electrons of N atoms form strong coordination bonds with Ce atoms. Considering that the coordination number of Ce is up to 12 in -CeN6, we deduce that electron transfer between the Ce atoms and N-structures play a crucial role in stabilizing -CeN6 at the relatively low synthesis pressure of 32 GPa.
Figure 3.(a) ELF of -CeN6 (isovalue = 0.8). (b) The charge transfer and formation energy of CeNn (n = 1, 2, 3, 4 and 6) compounds at 32 GPa.
To confirm our idea, Bader charges were calculated and clearly show the charge transfer for each Ce atoms and N-structures in stable CeNn (n = 1, 2, 3, 4, and 6) compounds at 32 GPa. The amount of charge transferred increases with nitrogen content from Fm-3m-CeN to C2/m-CeN3 [Fig. 3(b)], and the formation energy decreases from −4.37 to −5.83 eV/f.u., because the Ce atoms need to contribute a greater amount of charge in the N-rich phases than in the N-poor ones to stabilize the structure. As the nitrogen concentration continues to increase, the amount of charge transferred no longer increases, because the Ce atom reaches its highest valence. In spite of this, the excess nitrogen atoms can share electrons with others and form coordination bonds with Ce atoms, and hence the formation energy just increases from −5.83 eV/f.u. (C2/m-CeN3) to −5.67 eV/f.u. (-CeN6). Consequently, the donor electrons and the strong ligand effect of Ce atoms are two important factors stabilizing high-nitrogen compounds.
To determine the unique ligand effect of Ce in -CeN6, we performed a comparative analysis by simulating the high-pressure polymerization behavior of N2 molecules under two reaction environments. The projection of the COHP (pCOHP), the integral of the COHP (ICOHP), and the projected density of states (PDOS) were calculated to analyze the bonding features and bonding strength. According to the lattice parameters and stoichiometric ratio of -CeN6, we constructed a -N2 molecular crystal with six N atoms per cell, and made it more reasonable and stable by geometrical optimization at 32 GPa. The distance between the N2 molecules decreases dramatically to about 2.37–2.45 Å [Fig. 4(a)], but it is still larger than length of an N–N single bond (1.45 Å), suggesting that there is no bonding reaction between N2 molecules. The large −ICOHP (26.4) of the N–N bond and the strong hybridization interaction between N_2s and N_2p orbitals indicate strong covalent interaction in the N–N bond [Figs. 4(b) and 4(c)]. All the above points support the fact that the N≡N triple bonds in N2 molecules are hard to break down at 32 GPa.
Figure 4.(a) Crystal structure, (b) −pCOHP and −ICOHP, and (c) PDOS of N2 molecular crystal. (d) Crystal structure, (e) −pCOHP and −ICOHP, and (f) PDOS of trans-CeN6. (g) Crystal structure, (h) −pCOHP and −ICOHP, and (i) PDOS of -CeN6 at 32 GPa.
However, when we insert a Ce atom into the body-centered position (0.5, 0.5, 0.5) of -N2, the transitional CeN6 (trans-CeN6) with cis-form N chains is formed after optimization at the same pressure [Fig. 4(d)]. The hybridization interaction between N_2s and N_2p orbitals is weakened. The pCOHP of N–N bonds changes remarkably, and the −ICOHP value (12.9) of the N–N bonds is reduced by half compared with its value (26.4) in the absence of Ce atoms [Fig. 4(e)]. At this time, the N–N bond strength is close to that in P21–LiN5 (−ICOHP = 15.3) at 32 GPa,74 indicating that N≡N triple bonds have become N=N double or N–N single bonds. The reaction mechanism is similar to that in which Ce atoms are able to catalyze the production of NH3 from N2 through the breaking of N≡N triple bonds.2 Although the Ce–N bonds exhibit an ionic bond interaction (−ICOHP = 2.1), the interaction between Ce_4f and N_2p orbitals plays a crucial role in the reaction [Figs. 4(e) and 4(f)]. Besides, each Ce atom provides 2.10e to N2 molecules to assist their transformation to a polymerized N chain. More interestingly, the reaction Ce + 3N2 → trans-CeN6 is exothermic, with an energy release of 3.19 eV/f.u. at 32 GPa, indicating that the above process tends to occur spontaneously under high pressure.
Laser heating is an effective method to produce metal polynitrides under high pressure. As shown in Fig. 4(g), -CeN6 should be obtainable from trans-CeN6 by laser heating. Because the reaction trans-CeN6 → -CeN6 is exothermic, with an energy release of 2.48 eV/f.u., the high-temperature environment could increase the anharmonic vibration of atoms and accelerate the conversion of thermodynamically unstable trans-CeN6 into the stable phase -CeN6 at 32 GPa [Fig. S1(e) supplementary material]. Although the average −ICOHP of N–N bonds in -CeN6 (11.2) is a bit smaller than that in trans-CeN6 (12.9) [Fig. 4(h)], the total number of N–N bonds increases from six to seven in a unit cell, and the total −ICOHP increases from 77.2 to 78.50. The larger −ICOHP (2.7) of Ce–N bonds and the greater amount of charge transferred (2.20e) for the Ce atom in -CeN6 mean that the Ce–N interaction becomes stronger than that in trans-CeN6. As shown in Fig. 4(i), the PDOS near the Fermi level is remarkably different from that of trans-CeN6, indicating that a new atomic coordinated environment is formed in -CeN6, which exhibits a more stable configuration than that of trans-CeN6. According to Fig. S6 (supplementary material), -CeN6 is an insulator with a bandgap of 3.25 eV. It is obvious that the bandgap increases with increasing pressure, which is similar to the behavior of AlN4, AlN5, and N10 cages.11,51,75 The reason for this is that the strong coupling interaction of sp2 or sp3 orbitals in adjacent N atoms reduces the energy of the bonding state (valence band) and increases the energy of the antibonding state (conduction band), resulting in an increase in the gap.
C. Stability under ambient conditions
Quenching down the high-pressure phase to ambient conditions is of great significance for the practical application of HEDMs. Here, we further confirm the stability of -CeN6 under ambient conditions. The calculated phonon dispersion curve and elastic constants indicate that -CeN6 possess dynamical and mechanical stability at ambient pressure [Fig. 5(a) and Table SⅣ (supplementary material)]. An AIMD simulation was also performed. At 300 and 1000 K, the total energy of -CeN6 fluctuates about the equilibrium position and the nitrogen skeleton remains intact, indicating that this material possesses thermal stability under ambient conditions and 1000 K [Fig. S7 (supplementary material)]. In the radial distribution functions (RDFs), the first sharp peaks of each line demonstrate the nearest N–N, Ce–N, and Ce–Ce distances. The nearest N–N and Ce–N distances of -CeN6 are 1.44 and 2.59 Å at 0 GPa and 300 K, which are larger than those at 32 GPa and 300 K (1.41 and 2.47 Å, respectively) [Figs. 5(b) and S8 (supplementary material)]. The nearest N–N and Ce–N distances at 1000 K (1.43 and 2.58 Å) are comparable to those at 300 K. To sum up, -CeN6 can be quenched to ambient conditions if synthesized. In addition, C2/m-CeN3, -CeN3, and -CeN4 are also dynamically, mechanically, and thermally stable under ambient conditions [Figs. S7 and S9 and Table SⅣ (supplementary material)], and they can maintain thermal stability up to 1000, 700, and 400 K, respectively.
Figure 5.(a) Phonon dispersion curve and PHDOS of -CeN6 at 0 GPa. (b) RDFs g(r) and structures of final states from the last 2 ps of AIMD simulations at ambient pressure and temperatures of 300 K (solid lines) and 1000 K (dotted lines). (c) PDOS of -CeN6 at 0 GPa. (d) Curves of charge transfer and enthalpy as functions of pressure during pressure release in -CeN6, with the enthalpy of -CeN6 at 32 GPa being taken as the reference zero point.
Usually, high nitrogen content and stability under ambient conditions are mutually exclusive. According to the analysis above, the stability of -CeN6 in a high-pressure environment results from the high coordination number of Ce atoms and from charge transfer. Interestingly, as the pressure decreases to ambient, the PDOS shows that the DOS of N_2p orbitals in the valence band increases slightly and can hold more electrons [Fig. 5(c)]. The Bader charge analysis supports the same conclusions, with the amount of charge transferred increasing as the pressure decreases in -CeN6, indicating that the Ce atoms tend to provide more electrons at low pressure to enhance the stability of the N structure [Fig. 5(d)]. Meanwhile, the enthalpy decreases with decreasing pressure. Hence, charge transfer plays an important role in the stability of N structures at ambient pressure.
D. Energy density and explosive performance
The energy density, detonation velocity, and detonation pressure are important parameters to evaluate the explosive performance of an HEDM. Usually, these parameters are significantly related to the nitrogen content, the bonding types, and the mass of the coordination element. The mass energy density and volumetric energy density of -CeN6 are calculated using the dissociation path under ambient pressure: -CeN6 → CeN + N2. As shown in Table I, the mass energy density of -CeN6 is 2.00 kJ/g, which is comparable to that of the reported CNO (2.2 kJ/g)76 and LiN5 (2.72 kJ/g),74 and larger than that of C2/m-FeN6 (1.83 kJ/g),38-GdN6 (1.62 kJ/g),52 and Ibam-BaN10 (1.33 kJ/g).30 Additionally, the volumetric energy density of -CeN6 is as high as 11.20 kJ/cm3, which is larger than those of the high explosives TNT (7.05 kJ/cm3) and HMX (10.83 kJ/cm3) and those of some metal polynitrides (β-BeN4, γ-BeN4, GdN6, ReN8, BeN10, MgN10, BaN10, CaN10, YN10, GaN15, ScN15, and YN15) (3.46–10.95 kJ/cm3), and close to the reported maximum value in Be–N compounds (12.7 kJ/cm3).29–32,39,40
| Compound | ρ (g/cm3) | Ed (kJ/g) | Ev (kJ/cm3) | Vd (km/s) | Pd (GPa) |
|---|
| -CeN6 | 5.60 | 2.00 | 11.20 | 13.60 | 128.95 |
| TNT | 1.64a | 4.30b | 7.05c | 6.90d | 19.00a |
| HMX | 1.90a | 5.70b | 10.83c | 9.10d | 39.30a |
Table 1. Mass density ρ, mass energy density Ed, volumetric energy density Ev, detonation velocity Vd, and detonation pressure Pd of -CeN6 compared with those of TNT and HMX.
Interestingly, -CeN6 possesses outstanding detonation velocity Vd and detonation pressure Pd. Its detonation pressure is 128.95 GPa, which is about seven times that of TNT (19.00 GPa) and more than three times that of HMX (39.30 GPa), and its detonation velocity is 13.60 km/s, which is twice that of TNT (13.60 km/s). Moreover, the detonation pressure and velocity of -CeN6 are also greater than those of typical metal polynitrides, such as BeN4, ScN6, ScN7, GdN6, SnN20, and MN10 (M = Be, Mg, Ba, Ca, and Y) and MN15 (M = Al, Ga, Sc, and Y) compounds (15.81–100.96 GPa and 5.22–13.04 km/s).29–33,39,41 Notably, the excellent volumetric energy density, detonation pressure, and detonation velocity of -CeN6 are caused by its high mass density (up to 5.6 g/cm3). Thus, -CeN6 exhibits excellent explosive properties as a novel HEDM under ambient pressure.
E. Mechanical properties
Mechanical properties are important for the practical application of Ce polynitrides. Hence, the bulk modulus B, shear modulus G, Young’s modulus E, Poisson’s ratio υ, and Vickers hardness Hv of -CeN6 were calculated at ambient pressure (Table II). The C33 (469 GPa) of -CeN6 is greater than its C11 (273 GPa) and C22 (371 GPa), indicating greater incompressibility along the [001] directions than the [100] and [010] directions. The hardness of -CeN6 is 20.7 GPa, which is greater than those of AlN5 (15.2 GPa), MnN4 (17.5 GPa), HfN10 (13.7 GPa), NbN4 (17.9 GPa), ScN5 (17.4 GPa), ScN3 (17.5 GPa), M-ReN8, (13.9 GPa) and Tʹ-ReN8 (14.1 GPa), and comparable to those of FeN6 (24.9 GPa), RuN3 (23.4 GPa), and IrN4 (22.4 GPa),77–79 indicating that -CeN6 is a typical hard material (>20 GPa). The calculated Poisson’s ratio of -CeN6 is 0.2. This low Poisson’s ratio is the result of directional bonds, which increase the shear modulus and limit the movement of dislocations, increasing the hardness of the material.77 The small B/G ratio (<1.75) reveals that -CeN6 is a brittle material.
| B (GPa) | G (GPa) | E (GPa) | ν | Hv (GPa) | B/G |
|---|
| 155.00 | 118.12 | 282.59 | 0.20 | 20.73 | 1.31 |
Table 2. Bulk modulus B, shear modulus G, Young’s modulus E, Poisson’s ratio ν, and Vickers hardness Hv of -CeN6.
Additionally, the material stiffness of -CeN6 is anisotropic. The 3D surface of the Young’s modulus of -CeN6 is deformed [Fig. 6(a)], and the Young’s modulus of -CeN6 along the c axis is higher than that along the other direction owing to the strong N–N covalent bonds, indicating that the layered structure with novel N14 rings along the [001] direction is more incompressible [Fig. 6(b)]. The degree of crystal anisotropy can be described quantitatively by the ratio Emax/Emin. The Emax/Emin value of -CeN6 is 2.40, which is less than those of tr-FeN4 (3.74) and ReN8 (4.95–13.49),40 indicating a weaker crystal anisotropy.
Figure 6.(a) 3D surface and (b) 2D projected profiles of Young’s modulus in -CeN6.
F. IR and Raman spectra
The IR and Raman spectra of -CeN6 were calculated for experimental reference. From a group-theoretical analysis, the irreducible representation of -CeN6 is Γ = 12AuI + 9AgR. Therefore, -CeN6 has 18 vibrational modes after the removal of three acoustic modes (3AuI). In Fig. S10 (supplementary material), the modes of IR and Raman activity are denoted by I and R, respectively. The corresponding vibrational modes are presented in Figs. S11–S14 (supplementary material). -CeN6 contains 9 IR-active modes (9Au) and 9 Raman-active modes (9Ag). At 32 GPa, both the Au modes (274, 308, 355, 389, 509, 671, 683, 1009, and 1107 cm−1) and Ag modes (465, 550, 674, 811, 947, 1011, 1036, 1078, and 1169 cm−1) correspond to the out-of-plane N–N bending vibrations of folded N14 rings. At 0 GPa, both the Au modes (169, 215, 220, 278, 439, 605, 625, 859, and 1003 cm−1) and Ag modes (322, 481, 571, 699, 820, 875, 934, 967, and 1059 cm−1) also correspond to out-of-plane N–N bending vibration in the layered N-structure.
IV. CONCLUSION
A systematic high-pressure study of CeNn (n = 0.5, 1, 2, 3, 4, 5, and 6) compounds has been performed using first-principles swarm-intelligence structural searches. The phase diagram of CeNn compounds has been enriched by the proposal of six new stable high-pressure phases (I4/mmm-CeN2, C2/m-CeN3, -CeN3, -CeN4, C2/c-CeN6, and -CeN6). The stability of the new phases has been verified using the phonon dispersion curve, elastic constants, and AIMD simulations. The proposed layered structure of -CeN6 is composed of novel N14 ring. Not only does -CeN6 possess the lowest synthesis pressure of 32 GPa among the layered metal nitrides, but also it can be quenched down to ambient conditions. The reaction path Ce + 3N2 → trans-CeN6 → -CeN6 has been proposed to clarify the formation mechanism of -CeN6 under high pressure. The calculated results for COHP and electronic structure reveal that the charge transfer and orbital hybridization of Ce and N atoms play a crucial role in stabilizing -CeN6. The volumetric energy density of -CeN6 reaches 11.20 kJ/cm3, which is much higher than those of TNT and HMX. The detonation pressure (128.95 GPa) and detonation velocity (13.60 km/s) of -CeN6 are respectively almost seven times and twice those of TNT. Hence, -CeN6 has great application potential as an explosive material owing to its high stability and excellent explosive properties under ambient conditions.
SUPPLEMENTARY MATERIAL
ACKNOWLEDGMENTS
Acknowledgment. This work was supported financially by the National Key R&D Program of China (Grant Nos. 2018YFA0305900 and 2018YFA0703404), the National Natural Science Foundation of China under Grant Nos. 21905159, 11634004, 51320105007, 11604116, and 51602124, the Program for Changjiang Scholars and Innovative Research Team in the University of the Ministry of Education of China under Grant No. IRT1132, the Higher Educational Youth Innovation Science and Technology Program Shandong Province (Grant No. 2022KJ183), and GHfund B (Grant No. 202202026143).
References
[1] X. Q.Chen, C. L.Fu, R.Podloucky. Bonding and strength of solid nitrogen in the cubic gauche (cg-N) structure. Phys. Rev. B, 77, 064103(2008).
[2] Z.Pang, M.Sun, Y.Yin. Predicted new structures of polymeric nitrogen under 100–600 GPa. Comput. Mater. Sci., 98, 399-404(2015).
[3] T.Cui, D.Duan, X.Huang, X.Jin, B.Liu, F.Tian, L.Wang, X.Wang. Predicted novel metallic metastable phases of polymeric nitrogen at high pressures. New J. Phys., 15, 013010(2013).
[4] T.Cui, B.Liu, F.Tian, L.Wang, X.Wang, G.Zou. Structural stability of polymeric nitrogen: A first-principles investigation. J. Chem. Phys., 132, 024502(2010).
[5] S. V.Bondarchuk, B. F.Minaev. Two-dimensional honeycomb (A7) and zigzag sheet (ZS) type nitrogen monolayers. A first principles study of structural, electronic, spectral, and mechanical properties. Comput. Mater. Sci., 133, 122-129(2017).
[6] J.Hooper, A.Hu, T.Woo, F.Zahariev, F.Zhang. Layered single-bonded nonmolecular phase of nitrogen from first-principles simulation. Phys. Rev. B, 72, 214108(2005).
[7] J.Kotakoski, Z.Li, Y.Ma, A. R.Oganov, Y.Xie. Novel high pressure structures of polymeric nitrogen. Phys. Rev. Lett., 102, 065501(2009).
[8] T.Cui, Z.He, J. F.Li, B. B.Liu, Z. M.Liu, Y. M.Ma, X. L.Wang, G. T.Zou. Prediction of a new layered phase of nitrogen from first-principles simulations. J. Phys.: Condens. Matter, 19, 425226(2007).
[9] K.Albe, J.Kotakoski. First-principles calculations on solid nitrogen: A comparative study of high-pressure phases. Phys. Rev. B, 77, 144109(2008).
[10] S.Chiesa, R. M.Martin, W. D.Mattson, D.Sanchez-Portal. Prediction of new phases of nitrogen at high pressure from first-principles simulations. Phys. Rev. Lett., 93, 125501(2004).
[11] L.Chen, T.Cui, J.Li, J.Lv, Y.Ma, M.Miao, C. J.Pickard, X.Wang, Y.Wang, X.Zhong. Cagelike diamondoid nitrogen at high pressures. Phys. Rev. Lett., 109, 175502(2012).
[12] R.Boehler, D. A.Dzivenko, M. I.Eremets, A. G.Gavriliuk, I. A.Trojan. Single-bonded cubic form of nitrogen. Nat. Mater., 3, 558-563(2004).
[13] M.Kim, J.Smith, D.Tomasino, C. S.Yoo. Pressure-induced symmetry-lowering transition in dense nitrogen to layered polymeric nitrogen (LP-N) with colossal Raman intensity. Phys. Rev. Lett., 113, 205502(2014).
[14] G.Geneste, D.Laniel, P.Loubeyre, M.Mezouar, G.Weck. Hexagonal layered polymeric nitrogen phase synthesized near 250 GPa. Phys. Rev. Lett., 122, 066001(2019).
[15] S.Chariton, N.Dubrovinskaia, L.Dubrovinsky, T.Fedotenko, D.Laniel, V.Milman, A.Pakhomova, V.Prakapenka, B.Winkler. High-pressure polymeric nitrogen allotrope with the black phosphorus structure. Phys. Rev. Lett., 124, 216001(2020).
[16] G.Gaiffe, G.Garbarino, D.Laniel, P.Loubeyre, G.Weck. High-pressure synthesized lithium pentazolate compound metastable under ambient conditions. J. Phys. Chem. Lett., 9, 1600-1604(2018).
[17] M.Bykov, E.Bykova, S.Chariton, I.Chepkasov, A. F.Goncharov, E.Greenberg, S. Q.Jiang, A. R.Oganov, V. B.Prakapenka, A.Samtsevich, Y.Wang, X.Zhang. Stabilization of hexazine rings in potassium polynitride at high pressure. Nat. Chem., 14, 794-800(2022).
[18] M.Bykov, E.Bykova, N.Dubrovinskaia, L.Dubrovinsky, T.Fedotenko, E.Koemets, D.Laniel, B.Winkler. Synthesis of magnesium-nitrogen salts of polynitrogen anions. Nat. Commun., 10, 4515(2019).
[19] J. C.Crowhurst, I. I.Oleynik, V. B.Prakapenka, E.Stavrou, B. A.Steele, J. M.Zaug. High-pressure synthesis of a pentazolate salt. Chem. Mater., 29, 735-741(2016).
[20] A. N.Aslandukov, A. A.Aslandukova, S.Chariton, N.Dubrovinskaia, L. S.Dubrovinsky, T.Fedotenko, K.Glazyrin, D.Laniel, V. B.Prakapenka. High-pressure synthesis of the beta-Zn3N2 nitride and the alpha-ZnN4 and beta-ZnN4 polynitrogen compounds. Inorg. Chem., 60, 14594-14601(2021).
[21] A. I.Abrikosov, I. A.Abrikosov, T.Bin Masood, M.Bykov, S.Chariton, N.Dubrovinskaia, L.Dubrovinsky, T.Fedotenko, K.Glazyrin, A. F.Goncharov, M.Hanfland, I.Hotz, M. I.Katsnelson, D.Laniel, M. F.Mahmood, A. V.Ponomareva, V. B.Prakapenka, A. N.Rudenko, J. S.Smith, F.Tasnadi. High-pressure synthesis of Dirac materials: Layered van der Waals bonded BeN4 polymorph. Phys. Rev. Lett., 126, 175501(2021).
[22] Q.Wei, H.Yan, M.Zhang. Une xpected ground-state crystal structures and mechanical properties of transition metal pernitrides MN2 (M = Ti, Zr, and Hf). J. Alloys Compd., 774, 918-925(2019).
[23] H.Dong, X.Dong, X.Li, J. M.McMahon, A. R.Oganov, J.Zhang. Theoretical study of the crystal structure, stability, and properties of phases in the V-N system. Phys. Rev. B, 104, 134111(2021).
[24] J.Botana, L.Chen, T.Cui, J.Li, L.Liu, M.Miao, X.Wang, M.Zhang, H.Zhu. Polymerization of nitrogen in lithium azide. J. Chem. Phys., 139, 164710(2013).
[25] L.Chen, J.Li, H.Lin, X.Wang, H.Zhu. Polymerization of nitrogen in cesium azide under modest pressure. J. Chem. Phys., 141, 044717(2014).
[26] Q.Li, H.Wang, Z.Wu, K.Yin, M.Zhang, X.Zhang. Structural and electronic properties of sodium azide at high pressure: A first principles study. Solid State Commun., 161, 13-18(2013).
[27] J.Li, J.Lin, Q.Rui, F.Wang, Q.Wang, X.Wang. A novel square planar N42− ring with aromaticity in BeN4. Matter Radiat. Extremes, 7, 038401(2022).
[28] K.Hu, H.Li, B.Liu, S.Liu, S.Niu, X.Shi, P.Wang, D.Xu, Z.Yao, C.Zhai. Pressure-stabilized polymerization of nitrogen in manganese nitrides at ambient and high pressures. Phys. Chem. Chem. Phys., 24, 5738(2022).
[29] H.Gao, C.Liu, J.Sun, Q.Wu, K.Xia, J.Yuan, X.Zheng. Pressure-stabilized high-energy-density alkaline-earth-metal pentazolate salts. J. Phys. Chem. C, 123, 10205-10211(2019).
[30] J.Sun, J.Wu, K.Xia, J.Yuan. High-energy-density pentazolate salts: CaN10 and BaN10. Sci. China-Phys. Mech. Astron., 64, 218211(2020).
[31] P.Gao, K.Hao, S.Liu, W.Lu, J.Lv, M.Zhou. Pressure-stabilized high-energy-density material YN10. J. Phys.: Condens. Matter, 34, 135403(2022).
[32] H.Gao, C.Liu, J.Sun, Q.Wu, K.Xia, J.Yuan, X.Zheng. Predictions on high-power trivalent metal pentazolate salts. J. Phys. Chem. Lett., 10, 6166-6173(2019).
[33] G.Frapper, F.Guégan, R.Larhlimi, H.Valencia, B.Wang. Prediction of novel tin nitride SnxNy phases under pressure. J. Phys. Chem. C, 124, 8080-8093(2020).
[34] X.Chen, X.Liu, M.Miao, W.Yi, L.Zhao, Y.Zheng. Packing high-energy together: Binding the power of pentazolate and high-valence metals with strong bonds. Mater. Des., 193, 108820(2020).
[35] Y. L.Li, H. Q.Lin, Z.Zeng, J.Zhang. Pressure-induced planar N6 rings in potassium azide. Sci. Rep., 4, 4358(2014).
[36] H.Gao, C.Liu, J.Sun, H. T.Wang, K.Xia, D.Xing, J.Yuan. A novel superhard tungsten nitride predicted by machine-learning accelerated crystal structure search. Sci. Bull., 63, 817-824(2018).
[37] T.Cui, D.Duan, D.Li, F.Li, Z.Liu, F.Tian, Q.Zhuang. Formation mechanism of insensitive tellurium hexanitride with armchair-like cyclo-N6 anions. Commun. Chem., 3, 42(2020).
[38] P.Chen, F.Gao, N.Gong, H.Gou, H.Liu, T.Shen, R.Tian, B.Wan, L.Wu, Y.Yao. Prediction of stable iron nitrides at ambient and high pressures with progressive formation of new polynitrogen species. Chem. Mater., 30, 8476-8485(2018).
[39] H.Dong, Z.Mu, M.Wen, F.Wu, X.Xie, X.Zhang, X.Zhang. Pressure-induced high-energy-density BeN4 materials with nitrogen chains: First-principles study. J. Phys. Chem. C, 125, 25376-25382(2021).
[40] H.Gou, Y.Li, B.Liao, J.Sun, S.Sun, B.Wan, L.Wu, J.Xu, P.Zhou. Ultra-incompressibility and high energy density of ReN8 with infinite nitrogen chains. J. Mater. Sci., 56, 3814-3826(2020).
[41] Q.Chang, S.Chen, Y.Guo, Z.Liu, H.Sun, Y.Sun, S.Wei, G.Yin, Z.Yu. Polymerization of nitrogen in two theoretically predicted high-energy compounds ScN6 and ScN7 under modest pressure. New J. Phys., 24, 083015(2022).
[42] X.Li, H.Niu, A. R.Oganov, J.Zhang. Pressure-stabilized hafnium nitrides and their properties. Phys. Rev. B, 95, 020103(2017).
[43] G.Frapper, B.Huang, A. R.Oganov, S.Yu, Q.Zeng, L.Zhang. Emergence of novel polynitrogen molecule-like species, covalent chains, and layers in magnesium–nitrogen MgxNy phases under high pressure. J. Phys. Chem. C, 121, 11037-11046(2017).
[44] G.Frapper, B.Huang. Barium–nitrogen phases under pressure: Emergence of structural diversity and nitrogen-rich compounds. Chem. Mater., 30, 7623-7636(2018).
[45] J.Li, J.Lin, D.Peng, Q.Wang, X.Wang, H.Zhu. Stable nitrogen-rich scandium nitrides and their bonding features under ambient conditions. Phys. Chem. Chem. Phys., 23, 6863-6870(2021).
[46] H.Li, B.Liu, R.Liu, S.Liu, X.Shi, P.Wang, Z.Yao. Cobalt-nitrogen compounds at high pressure. Inorg. Chem., 60, 14022-14030(2021).
[47] H.Li, Z.Li, B.Liu, S.Niu, X.Shi, Z.Yao. New cadmium-nitrogen compounds at high pressures. Inorg. Chem., 60, 6772-6781(2021).
[48] I. I.Oleynik, B. A.Steele. Novel potassium polynitrides at high pressures. J. Phys. Chem. A, 121, 8955-8961(2017).
[49] K.Bao, T.Cui, D.Duan, D.Li, B.Liu, Z.Liu, F.Tian, W.Wang, S.Wei. A novel polymerization of nitrogen in beryllium tetranitride at high pressure. J. Phys. Chem. C, 121, 9766-9772(2017).
[50] X.Dong, G.Gao, J.Hou, A. R.Oganov, X.Shao, Y.Tian, H. T.Wang, X. J.Weng, X. F.Zhou. Helium-nitrogen mixtures at high pressure. Phys. Rev. B, 103, L060102(2021).
[51] T.Cui, D.Duan, D.Li, Y.Liu, Z.Liu, F.Tian. Metallic and anti-metallic properties of strongly covalently bonded energetic AlN5 nitrides. Phys. Chem. Chem. Phys., 21, 12029(2019).
[52] L.Liu, D.Wang, H.Zhang, S.Zhang. Pressure-stabilized GdN6 with an armchair–antiarmchair structure as a high energy density material. J. Mater. Chem. A, 9, 16751-16758(2021).
[53] H.Cai, K.Chen, Y.Feng, X.Jiang, X.Wang, J.Zeng, Y.Zheng. Prediction of erbium–nitrogen compounds as high-performance high-energy-density materials. J. Phys.: Condens. Matter, 35, 085701(2023).
[54] S.Chen, Y.Fu, S.Guo, X.Hu, J.Sun, Y.Sun, S.Zhang, J.Zhu. Identifying electrocatalytic activity and mechanism of Ce1/3NbO3 perovskite for nitrogen reduction to ammonia at ambient conditions. Appl. Catal., B, 280, 119419(2021).
[55] S.Lin, J.Qi, K.Xie, S.Zhou. Catalytic role of assembled Ce Lewis acid sites over ceria for electrocatalytic conversion of dinitrogen to ammonia. J. Energy Chem., 60, 249-258(2021).
[56] H.Chen, G.Cui, F.Gong, Q.Liu, X.Sun, T.Wang, L.Xia, X.Xiong, B.Xu, R.Zhao, F.Zhou, Q.Zhou. Enhancing electrocatalytic N2 reduction to NH3 by CeO2 nanorod with oxygen vacancies. ACS Sustainable Chem. Eng., 7, 2889-2893(2019).
[57] O.Eriksson, V.Kanchana, Y.Ma, A.Svane, G.Vaitheeswaran, X.Zhang. Lattice dynamics and elastic properties of the 4f electron system: CeN. Phys. Rev. B, 84, 205135(2011).
[58] M.Bremholm, D.Ceresoli, J.-E.J?rgensen, M. B.Nielsen, V. B.Prakapenka, C.Prescher. Experimental evidence for pressure-induced first order transition in cerium nitride from B1 to B10 structure type. J. Appl. Phys., 121, 025903(2017).
[59] H.Wang, Q.Wei, H.Yan, M.Zhang. Exploration on pressure-induced phase transition of cerium mononitride from first-principles calculations. Appl. Phys. Lett., 102, 231901(2013).
[60] J.Lv, Y.Ma, Y.Wang, L.Zhu. CALYPSO: A method for crystal structure prediction. Comput. Phys. Commun., 183, 2063-2070(2012).
[61] J. F. l. G.Kresse. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B, 54, 11169-11186(1996).
[62] K.Burke, M.Ernzerhof, J. P.Perdew. Generalized gradient approximation made simple. Phys. Rev. Lett., 77, 3865-3868(1996).
[63] J.Bai, A. M.Hao. First-principles calculations of electronic and magnetic properties of CeN: The LDA + U method. Chin. Phys. B, 22, 107102(2013).
[64] Y. X.Wang, G. B.Zhang, Y. G.Zhang. First-principles study of the electronic structure and optical properties of Ce-doped ZnO. J. Appl. Phys., 109, 063510(2011).
[65] R. A.De Souza, M.Martin, A.Schriever, T.Zacherle. Ab initio analysis of the defect structure of ceria. Phys. Rev. B, 87, 134104(2013).
[66] C. W. M.Castleton, K.Hermansson, J.Kullgren. Tuning for electron localization and structure at oxygen vacancies in ceria. J. Chem. Phys., 127, 244704(2007).
[67] D. J. G.Kresse. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B, 59, 1758-1775(1999).
[68] A. V.Krukau, J. P.Perdew, A.Savin, G. E.Scuseria. Hybrid functionals with local range separation. J. Chem. Phys., 129, 124103(2008).
[69] L.Chaput, I.Tanaka, A.Togo. Distributions of phonon lifetimes in Brillouin zones. Phys. Rev. B, 91, 094306(2015).
[70] M. J. K. a. C.Dickinson. Evaluation of the simplied calculational method for Chapman-Jouguet detonation pressures on the basis of available experimental information. J. Chem. Phys., 48, 43-50(1968).
[71] M.Parrinello, A.Rahman. Crystal structure and pair potentials: A molecular-dynamics study. Phys. Rev. Lett., 45, 1196-1199(1980).
[72] V. L.Deringer, R.Dronskowski, A. L.Tchougreeff. Crystal orbital Hamilton population (COHP) analysis as projected from plane-wave basis sets. J. Phys. Chem. A, 115, 5461-5466(2011).
[73] S. J.Clark, P. J.Hasnip, P. J. D.Lindan, M. C.Payne, C. J.Pickard, M. J.Probert, M. D.Segall. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys.: Condens. Matter, 14, 2717-2744(2002).
[74] H.Liu, Y.Ma, F.Peng, Y.Yao. Crystalline LiN5 predicted from first-principles as a possible high-energy material. J. Phys. Chem. Lett., 6, 2363-2366(2015).
[75] T.Gao, S.Li, S.Ma, F.Peng, S.Zhu. Novel phase of AlN4 as a possible superhard material. J. Phys. Chem. C, 122, 22660-22666(2018).
[76] C. J.Pickard, C.Pinilla, Z.Raza, A. M.Saitta. High energy density mixed polymeric phase from carbon monoxide and nitrogen. Phys. Rev. Lett., 111, 235501(2013).
[77] K.Bao, T.Cui, D.Duan, B.Liu, F.Tian, Z.Zhao. Phase diagram, mechanical properties, and electronic structure of Nb-N compounds under pressure. Phys. Chem. Chem. Phys., 17, 22837-22845(2015).
[78] R.Gao, H.Gou, Q.Hu, Z.Li, Y.Lin, B.Wan, L.Wu, J.Zhang, Y.Zhang, Y.Zhao. Diverse ruthenium nitrides stabilized under pressure: A theoretical prediction. Sci. Rep., 6, 33506(2016).
[79] X.Du, J.Wang, G.Yang, Q.Yang, Y.Yao. IrN4 and IrN7 as potential high-energy-density materials. J. Chem. Phys., 154, 054706(2021).