
Jianan Yuan, Kang Xia, Chi Ding, Xiaomeng Wang, Qing Lu, Jian Sun. High-energy-density metal nitrides with armchair chains[J]. Matter and Radiation at Extremes, 2022, 7(3): 038402
- Matter and Radiation at Extremes
- Vol. 7, Issue 3, 038402 (2022)
Abstract
I. INTRODUCTION
Molecular nitrogen (N2) is the most abundant component of Earth’s atmosphere and one of the most stable molecules owing to its strong N≡N triple bond. There is a large energy difference between single/double nitrogen bonds (160 and 418 kJ/mol for N–N and N=N, respectively) and the N≡N triple bond (954 kJ/mol).1 Therefore, polynitrogen composed of mixed single and double bonds can store a substantial amount of chemical energy. The stored energy will be released when decomposition of the polymeric nitrogen is triggered, producing pollution-free N2 molecules, in an oxygen-free process. However, the high bond energy of the N2 molecule makes it highly unreactive. It is difficult to synthesize polymeric nitrogen from N2 molecules. To break nitrogen triple bonds and obtain nitrogen polymers, extreme conditions such as high pressures and high temperatures become necessary to overcome the high energy barriers.2,3 For example, pressure can be used to break the nitrogen bonds, with the formation of some unexpected nitrogen compounds.4 In some nitrogen-rich cases, pressure can enhance the effect of electronic delocalization, thus helping to dissociate molecules and form polymeric structures.
Tremendous efforts have been made to obtain novel polymeric nitrogen structures under high pressures in both theoretical and experimental studies.2,3,5–9 More than a decade after its theoretical prediction by Mailhiot et al.,5 an experimental breakthrough was eventually made by Eremets et al. in 2004 in their synthesis of a cubic-gauche nitrogen structure (cg-N).2 This cg-N contains pure N–N single bonds and thus possesses an extremely high energy density, which is reported to be five times higher than that of the current most powerful energetic materials.5 Since then, several polymeric nitrogen structures have been synthesized: hexagonal layered polymeric nitrogen (HLP-N), layered polymeric nitrogen (LP-N), and a black phosphorus structure (BP-N).10–13 However, the pressure–temperature conditions required for these polymeric nitrogen syntheses are very high, exceeding 110 GPa and 2000 K. Such extreme conditions cause great practical difficulties, and therefore the search for potential high-energy-density materials (HEDMs) that can be synthesized under moderate pressures is still an important task.
Recently, counter-ions and nitrogen fragments have been used to construct nitrogen-bearing compounds involving ionic bonding, including N5 and N6 rings, nitrogen azide, and polymeric N4 chains (poly-).7,14–28 In these cases, the ionic bonds can enhance the kinetic stability of the polynitride components and reduce the pressure required for synthesis. Among these nitrogen fragments, poly- has become of particular importance. Previous theoretical works have predicted the existence of several poly- compounds, including BeN4, MgN4, CaN4, CdN4, and FeN4.29–33 Of these, MgN4, FeN4, TaN4, and BeN4 have recently been synthesized.34–37 Typical alkaline-earth nitrides such as the BeN4, MgN4, and CaN4 compounds,29,30,32 have similar N–N bond lengths in the range 1.32–1.35 Å. However, the poly- chains in FeN434 possess significantly different N–N bond lengths, between 1.29 and 1.43 Å. Nevertheless, these poly- chains are constructed with N–N single bonds and N=N double bonds, matching the features of HEDM nitrides. Additionally, the species is found to coexist with the poly- chain in some high-energy-density metal nitrides synthesized by Bykov et al.38,39 The cations in these structures are mainly divalent metals. We envision that the nitrogen content of metal nitrides can be increased by the use of metal ions in higher valence states, thereby achieving higher energy densities.
In this work, we focus on the metals Al, Ga, Y and Ti, which can provide three or four valence electrons. We predict that the poly- chain can occur in four new metal nitride crystals MNx with different symmetries: P21-AlN6, P21-GaN6, P-1-YN6, and P4/mnc-TiN8. These are predicted to be energetically stable at moderate pressures. Molecular dynamics (MD) simulations and phonon spectrum calculations suggest that these MNx are mechanically stable at nonzero temperatures and high pressures. More interestingly, we find coexistence of the aforementioned two types of poly- chains, appearing in our predicted P21 phase of the AlN6 and GaN6 compounds. Besides, we estimate that these MNx have excellent detonation properties and can served as potential HEDMs.
II. METHODS
We carry out a crystal structure search using MAGUS (“machine learning and graph theory assisted universal structure searcher”),40,41 which is accelerated through the use of graph theory42 and machine learning potentials. This method has been successfully applied to many systems.40,43–48 To generate new structures containing the desired units, molecular-based unit searches are conducted at 50 GPa. We constrain four MNx formula units into one cell to explore more nitrogen-containing configurations. The first-principles calculations are performed using the Vienna Ab initio Simulation Package (VASP).49 Density-functional theory (DFT) is implemented within the projector-augmented-wave (PAW) approach.50 We use the Perdew–Burke–Erzernhof functional based on the generalized gradient approximation51,52 to describe the exchange–correlation interaction. Van der Waals (vdW) effects are taken into account using the DFT-D3 method in VASP.53,54 The valence electrons occupying the shell states are 3s23p1, 4s24p1, 4s24p65s14d2, and 3p64s23d2 for Al, Ga, Y, and Ti, respectively. The energy cutoff of the plane-wave basis is set at 1050 eV. The Brillouin zone is separated with k-meshes of 2π × 0.03 Å−1 to guarantee convergence of energy. We use the PHONOPY code55 to calculate the phonon spectrum for a 2 × 2 × 2 supercell. Projected crystal orbital Hamilton population (pCOHP) calculations are conducted using the LOBSTER code.56,57 Noncovalent interactions are also taken into consideration by calculating the reduced density gradient (RDG) using the Critic2 code.58,59 The structures are visualized by VESTA software.60 We analyze the electronic properties in the context of molecular orbital (MO) theory.61
III. RESULTS AND DISCUSSION
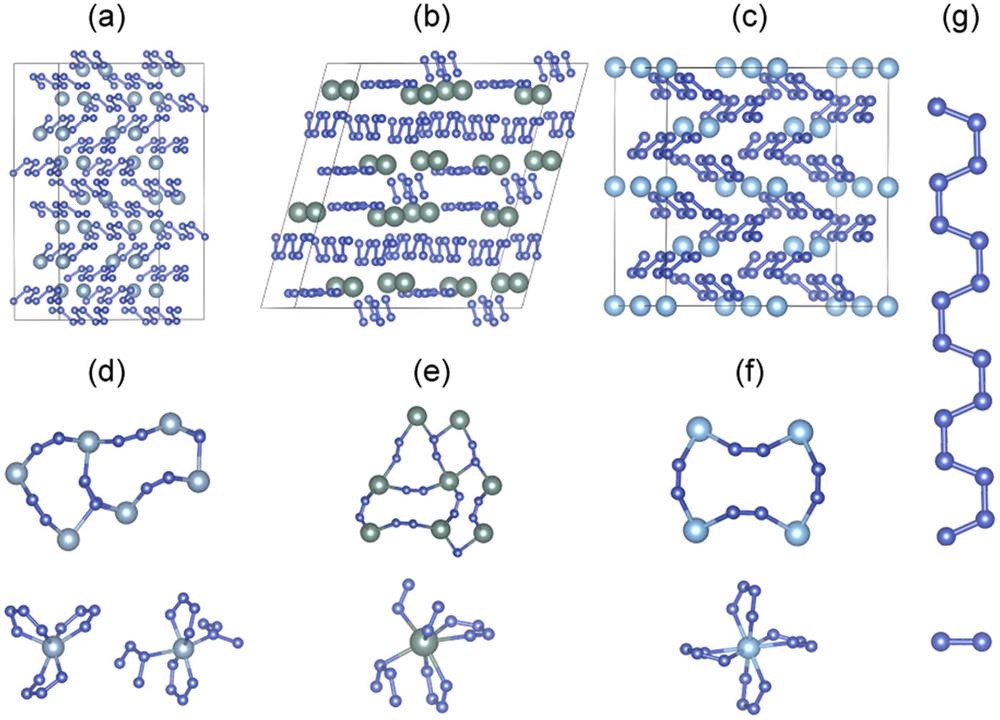
Using the crystal structure search method described above, we predict several metal nitrides structures, as shown in Figs. 1(a)–1(c). Diverse channel frames are constructed from different coordinate bonds between metal atoms and poly- chains [Figs. 1(d)–1(f)]. These channel-like structures may capture small molecules, such as N2 or H2O, all of which stabilize these metal nitrides.17,18,39 For AlN6 and GaN6 in the P21 phase, one metal atom is found to be coordinated with three or four chains of poly- [Fig. 1(d)]. As shown in Fig. 1(e), one metal atom in the P-1-YN6 structure is sevenfold-coordinated with nitrogen atoms. Because N2 double balls coexist with poly- chains in the P-1-YN6 structure, we can describe the YN6 within a unit cell as . For P4/mnc-TiN8, the poly- chain is fourfold-coordinated around one Ti atom [Fig. 1(f)].
![]()
Figure 1.(a) Isostructural AlN6 and GaN6 of the
We calculate the relative enthalpy of MNx (M = Al, Ga, Y, and Ti) as shown in Fig. 2. By taking the enthalpy difference of MNx relative to the ground-state metal nitrides (MN) plus bulk nitrogen phase at these pressures, ΔH = MNx − MN − (x − 1)N, we can confirm the energetic stabilities of MNx compounds under pressure. Here, the Pa-3 (α) and P41212 phases of the N2 molecular crystal structures are selected for the relative-enthalpy calculations at 0 GPa and at higher pressures, respectively.65Fm-3m-MN is chosen as the ground state for the Al–N, Y–N and Ti–N systems. The ground state for the GaN structure in the Ga–N system favors the P63/mmc phase, which tends to transform into the Fm-3m phase above 32.5 GPa.62 We find that AlN6, GaN6, and TiN8 are enthalpically stable above pressures of around 40.8, 39.3, and 40.1 GPa, respectively. However, YN6 turns out to be stable above a much lower external pressure (around 21.7 GPa). Therefore, we suggest that MNx might be synthesized at moderate pressures around 40 GPa or lower. In addition, no imaginary frequency of the phonon spectrum has been found in the Brillouin zones of these MNx structures, indicating their dynamical stability, as shown in Fig. S1 in the
![]()
Figure 2.Enthalpy difference of MN
To confirm the thermal stabilities of the newly predicted MNx, we conduct ab initio molecular dynamics (AIMD) simulations for the MNx (2 × 2 × 2) supercells at high temperatures for 12 ps with an interval of 1 fs. The supercells contain 224 atoms for the AlN6, GaN6, and YN6 structures and 144 atoms for TiN8. The AIMD calculations are performed within the canonical ensemble for AlN6 and GaN6 at 20 GPa, for YN6 at 40 GPa, and for TiN8 at 30 GPa. The resulting radial distribution functions (RDFs) are presented in Fig. 3 and in Fig. S2 in the
![]()
Figure 3.(a)–(d) Radial distribution functions (RDFs)
The calculated density of states (DOS) exhibits metallic features for all MNx structures, as shown in Figs. 4(a)–4(d). The main contribution to the DOS at the Fermi energy is from the N-2p orbital for the AlN6 and GaN6 structures, while the d orbitals of the transition metals Y and Ti make additional contributions to the DOS around the Fermi energy for YN6, and TiN8. We carry out a projected crystal orbital Hamilton population (pCOHP) calculation to distinguish the bond features in the predicted MNx nitrides. Using minus pCOHP (−pCOHP), we can partition the bond energy into bonding states with positive values and antibonding states with negative values. Figures 4(e)–4(h) reveal similar bond features for these four structures. For N–N bonds, although most of the states below the Fermi level can be attributed to bonding states, there are still some antibonding states existing just below the Fermi level. For M–N bonds, however, almost all the states below the Fermi level are bonding states. The minus integral pCOHP (−IpCOHP)66 values at the Fermi level, which represent the pairwise interatomic interaction strength, are listed in Table I. The −IpCOHP values of N–N bonds are at least twice those of M–N bonds, which suggests that the N–N bonds have greater pairwise interatomic interaction strengths than the M–N bonds.
![]()
Figure 4.(a)–(d) Density of states (DOS) and (e)–(h) minus projected crystal orbital Hamilton population (−pCOHP) for
| Average −IpCOHP/bonds (eV/bond) | ||
|---|---|---|
| Compound | M–N | N–N |
| P21-AlN6 | 4.83 | 14.26 |
| P21-GaN6 | 4.28 | 14.51 |
| P-1-YN6 | 3.12 | 15.30 |
| P4/mnc-TiN8 | 2.72 | 12.46 |
Table 1. Average −IpCOHP values for MNx.
To explain the electronic features of MNx, we apply MO theory61 to analyze the electron orbitals and describe the coordination of the metal atoms. For the isostructural AlN6 and GaN6, two types of poly- chains (types A and B) are sketched in Figs. 5(a) and 5(b), respectively. Each N atom in type A [Fig. 5(a)] is surrounded by three atoms in a nearly planar triangular geometry, suggesting sp2 hybridization. Of a total of 22 electrons in one unit, 16 are found to occupy eight sp2 hybrid orbitals. The extra four p orbitals of four nitrogen atoms form four π orbitals. The remaining electrons occupying π orbitals form delocalized π bonds, which is the origin of metallicity. The equally distributed π band also explains why the N–N bond lengths (1.32–1.33 Å) are between those of an N–N single bond (1.45 Å) and an N=N double bond (1.20 Å).29 As shown in Fig. 5(b), for the type B poly- chain, the two N atoms colored red possess tetrahedral coordination including lone pairs, indicating their sp3 hybridization, while the other two N atoms are sp2-hybridized, similar to those in Fig. 5(a), and therefore the extra two p atomic orbitals of these two N atoms form one bonding orbital π and one antibonding orbital π*. The remaining two electrons occupying the bonding π orbital form a π bond, distributed over the sp2-hybridized N atoms. This can explain why the length of the bond between the two sp3-hybridized N atoms is 1.40 Å, which is close to that of an N–N single bond (1.45 Å), but the length of the bond connecting the sp2-hybridized N atoms is 1.30 Å, which is between those of single and double bonds. Therefore, the N–N bonds in the type A chain are all hybridized bonds, while those in the type B chain are half-single, half-hybridized. In terms of bond energy, the type B chain may possess a higher energy capability in principle. However, YN6 and TiN8 contain only type A poly- chains.
![]()
Figure 5.Sketches of (a) type A and (b) type B chains for N4–metal coordination, and fragment structures of (c) type A and (d) type B poly-
When the electron density ρ is too low to allow analysis of the results for the electron localization function, we can identify the noncovalent interactions (NCIs) by calculating the reduced density gradient (RDG) s, which is defined as
![]()
Figure 6.Plots of the RDG
Nitrides containing poly- chains have been synthesized for potential applications as HEDMs.34–37 This inspires us to study the detonation performance of the MNx structures predicted in this work, which have more poly- chains than the previously proposed structures. We assume that MNx will decompose into MN and N2 gas under ambient conditions: . The estimated detonation properties of MNx are listed in Table II. In particular, the energy densities of MNx exceed that of TNT (4.30 kJ/g), except for P21-GaN6 and P-1-YN6. We calculate their detonation pressures P and detonation velocities V using the Kamlet–Jacobs empirical equations22,69 and , where N is the number of moles of N2 gas generated during the decomposition of 1 g of MNx and M is the molar mass of N2 (28 g/mol). As can be seen from Table II, MNx have excellent detonation pressures and detonation velocities that are higher than those of the explosives TNT and HMX.
| ρ (g/cm3) | Eg (kJ/g) | Ev (kJ/cm3) | V (km/s) | P (kbar) | |
|---|---|---|---|---|---|
| AlN6 | 1.93 | 4.41 | 8.54 | 10.30 | 462 |
| GaN6 | 3.48 | 3.94 | 13.75 | 10.38 | 1020 |
| YN6 | 4.00 | 2.71 | 10.86 | 12.67 | 993 |
| TiN8 | 2.91 | 4.50 | 13.10 | 13.43 | 1024 |
| TNT | 1.64 | 4.30 | 7.05 | 6.90 | 190 |
| HMX | 1.90 | 5.70 | 10.83 | 9.10 | 393 |
Table 2. Comparison of the detonation properties of MNx structures estimated using the Kamlet–Jacobs empirical equations
IV. CONCLUSIONS
By applying a structure search method accelerated by machine learning, we have successfully predicted four metal nitrides constructed from poly- chains: P21-AlN6, P21-GaN6, P-1-YN6, and P4/mnc-TiN8. Based on first-principles calculations, we have found that the pressures required for synthesis of P21-AlN6, P21-GaN6, and P4/mnc-TiN8 are around 40 GPa, and that required for P-1-YN6 is near 20 GPa. Phonon calculations suggest that P4/mnc-TiN8 is mechanically stable at ambient pressure, while the other three structures are mechanically stable at high pressures. Furthermore, the dynamical stabilities of MNx at nonzero temperature have been shown by molecular dynamics simulations. Two types of poly- chains are found: in type A, all the N atoms are sp2-hybridized and form delocalized π bonds distributed along the entire chain; in type B, half of the nitrogen atoms are sp3-hybridized, while the other half adopt sp2 hybridization, and so π bonding can only occur for some of the N–N bonds in the poly- chain. The results for DOS and pCOHP also confirm the MO analysis. The metallicity of MNx stems from the delocalized π bonds. RDG calculations reveal that the effect of NCIs also helps to stabilize these MNx structures. Most importantly, we estimate that the newly predicted MNx have good detonation properties with potential applications as HEDMs. Furthermore, our predictions here can provide some guidance for future experiments.
SUPPLEMENTARY MATERIAL
ACKNOWLEDGMENTS
Acknowledgment. J.S. gratefully acknowledges financial support from the National Natural Science Foundation of China (Grant Nos. 12125404, 11974162, and 11834006), and the Fundamental Research Funds for the Central Universities. K.X. acknowledges support from the National Natural Science Foundation of China under Grant No. 12004185, the Natural Science Foundation of the Jiangsu Higher Education Institutions of China under Grant No. 20KJB140016, and the Scientific Research Start-up Funds of Nanjing Forestry University (No. 163101110), and financial support from a Project funded by the China Postdoctoral Science Foundation (Grant No. 2019M651767). The calculations were carried out using supercomputers at the High Performance Computing Center of the Collaborative Innovation Center of Advanced Microstructures, the High-Performance Supercomputing Center of Nanjing University, and the High-Performance Computing Facility of Nanjing Forestry University.
References
[1] D. P.Stevenson. The strengths of chemical bonds. J. Am. Chem. Soc., 77, 2350(1955).
[2] I. A.Trojan, A. G.Gavriliuk, R.Boehler, M. I.Eremets, D. A.Dzivenko. Single-bonded cubic form of nitrogen. Nat. Mater., 3, 558-563(2004).
[3] V. B.Prakapenka, J. M.Zaug, I. I.Oleynik, B. A.Steele, E.Stavrou, J. C.Crowhurst. High-pressure synthesis of a pentazolate salt. Chem. Mater., 29, 735-741(2017).
[4] M.Miao, E.Zurek, Y.Sun, H.Lin. Chemistry under high pressure. Nat. Rev. Chem., 4, 508-527(2020).
[5] L. H.Yang, C.Mailhiot, A. K.McMahan. Polymeric nitrogen. Phys. Rev. B, 46, 14419-14435(1992).
[6] K. O.Christe, J. A.Sheehy, W. W.Wilson, V.Vij, A.Vij, F. S.Tham. Polynitrogen chemistry. Synthesis, characterization, and crystal structure of surprisingly stable fluoroantimonate salts of
[7] G.Garbarino, G.Weck, P.Loubeyre, G.Gaiffe, D.Laniel. High-pressure synthesized lithium pentazolate compound metastable under ambient conditions. J. Phys. Chem. Lett., 9, 1600-1604(2018).
[8] Y.Ma, W.Lei, H.Liu, D.Liu, Y.Li, X.Feng, J.Hao, S. A. T.Redfern. Route to high-energy density polymeric nitrogen
[9] Y.Zhang, J.Sun, E.Greenberg, N. P.Salke, J. F.Lin, S.Fu, K.Xia, V. B.Prakapenka, J.Liu. Tungsten hexanitride with single-bonded armchairlike hexazine structure at high pressure. Phys. Rev. Lett., 126, 065702(2021).
[10] C.-S.Yoo, M.Kim, D.Tomasino, J.Smith. Pressure-induced symmetry-lowering transition in dense nitrogen to layered polymeric nitrogen (LP-N) with colossal Raman intensity. Phys. Rev. Lett., 113, 205502(2014).
[11] G.Geneste, D.Laniel, G.Weck, M.Mezouar, P.Loubeyre. Hexagonal layered polymeric nitrogen phase synthesized near 250 GPa. Phys. Rev. Lett., 122, 066001(2019).
[12] D.Laniel, V.Prakapenka, S.Chariton, B.Winkler, L.Dubrovinsky, T.Fedotenko, V.Milman, A.Pakhomova, N.Dubrovinskaia. High-pressure polymeric nitrogen allotrope with the black phosphorus structure. Phys. Rev. Lett., 124, 216001(2020).
[13] J. S.Smith, Y.Yao, C.Ji, B.Wan, A. A.Adeleke, W. L.Mao, H. K.Mao, Y.Meng, V. B.Prakapenka, L.Yang, G.Shen, W.Liu, B.Li, H.Gou. Nitrogen in black phosphorus structure. Sci. Adv., 6, eaba9206(2020).
[14] F.Peng, Y.Ma, H.Liu, Y.Yao. Crystalline LiN5 predicted from first-principles as a possible high-energy material. J. Phys. Chem. Lett., 6, 2363-2366(2015).
[15] Y.Yao, F.Peng, H.Liu, S.Zhu, T.Gao, A.Majumdar. Stable calcium nitrides at ambient and high pressures. Inorg. Chem., 55, 7550-7555(2016).
[16] B. A.Steele, I. I.Oleynik. Sodium pentazolate: A nitrogen rich high energy density material. Chem. Phys. Lett., 643, 21-26(2016).
[17] M.Lu, B.Hu, C.Yu, C.Sun, C.Zhang. Synthesis and characterization of the pentazolate anion
[18] Q.Lin, Q.Wang, C.Shen, P.Wang, M.Lu, Y.Xu. A series of energetic metal pentazolate hydrates. Nature, 549, 78-81(2017).
[19] S.Wei, L.Lian, B.Liu, P.Hou, B.Wang, D.Li, Y.Cai. Structural phase transition and bonding properties of high-pressure polymeric CaN3. RSC Adv., 8, 4314-4320(2018).
[20] G.Frapper, B.Huang. Barium–nitrogen phases under pressure: Emergence of structural diversity and nitrogen-rich compounds. Chem. Mater., 30, 7623-7636(2018).
[21] Z.Liu, Y.Liu, D.Duan, D.Li, T.Cui, F.Tian. Metallic and anti-metallic properties of strongly covalently bonded energetic AlN5 nitrides. Phys. Chem. Chem. Phys., 21, 12029-12035(2019).
[22] K.Xia, J.Sun, H.Gao, J.Yuan, C.Liu, X.Zheng, Q.Wu. Pressure-stabilized high-energy-density alkaline-earth-metal pentazolate salts. J. Phys. Chem. C, 123, 10205-10211(2019).
[23] Q.Wu, J.Sun, C.Liu, H.Gao, X.Zheng, K.Xia, J.Yuan. Predictions on high-power trivalent metal pentazolate salts. J. Phys. Chem. Lett., 10, 6166-6173(2019).
[24] M.Zhang, S.Zhang, Y.Tian, D.Zhang, M.Lu, T.Bi, X.Xu, L.Gao, Y.Du, Y.Yan. Predicted crystal structures of titanium nitrides at high pressures. Comput. Mater. Sci., 180, 109720(2020).
[25] B.Liu, Z.Yao, X.Shi. New high pressure phases of the Zn–N system. J. Phys. Chem. C, 124, 4044-4049(2020).
[26] R.Larhlimi, F.Guégan, G.Frapper, B.Wang, H.Valencia. Prediction of novel tin nitride Sn
[27] H.Zhang, C.Niu, J.Zhang, Z.Zeng, J.Zhao, X.Wang. Polymerization of nitrogen in nitrogen–fluorine compounds under pressure. J. Phys. Chem. Lett., 12, 5731-5737(2021).
[28] J.Wu, K.Xia, J.Yuan, J.Sun. High-energy-density pentazolate salts: CaN10 and BaN10. Sci. China: Phys., Mech. Astron., 64, 218211(2021).
[29] A. R.Oganov, B.Huang, G.Frapper, L.Zhang, Q.Zeng, S.Yu. Emergence of novel polynitrogen molecule-like species, covalent chains, and layers in magnesium–mitrogen Mg
[30] L.Liu, S.Zhang, Z.Zhao, G.Yang. Pressure-induced stable BeN4 as a high-energy density material. J. Power Sources, 365, 155-161(2017).
[31] Y.Yao, P.Chen, R.Tian, N.Gong, F.Gao, H.Liu, T.Shen, B.Wan, L.Wu, H.Gou. Prediction of stable iron nitrides at ambient and high pressures with progressive formation of new polynitrogen species. Chem. Mater., 30, 8476-8485(2018).
[32] Z.Yao, B.Liu, B.-B.Liu, X.-H.Shi. Pressure-stabilized new phase of CaN4. Chin. Phys. Lett., 37, 047101(2020).
[33] H.Li, Z.Li, S.Niu, Z.Yao, B.Liu, X.Shi. New cadmium–nitrogen compounds at high pressures. Inorg. Chem., 60, 6772-6781(2021).
[34] E.Koemets, K.Glazyrin, V.Prakapenka, M.Mezouar, C.McCammon, E.Bykova, F.Tasnádi, H. P.Liermann, N.Dubrovinskaia, G.Aprilis, M.Bykov, L.Dubrovinsky, A. V.Ponomareva, I.Kupenko, I.Chuvashova, I. A.Abrikosov. Fe-N system at high pressure reveals a compound featuring polymeric nitrogen chains. Nat. Commun., 9, 2756(2018).
[35] L.Dubrovinsky, D.Laniel, B.Winkler, M.Bykov, E.Koemets, N.Dubrovinskaia, E.Bykova, T.Fedotenko. Synthesis of magnesium-nitrogen salts of polynitrogen anions. Nat. Commun., 10, 4515(2019).
[36] F.Tasnádi, A. N.Rudenko, I.Hotz, K.Glazyrin, M. F.Mahmood, J. S.Smith, M.Bykov, N.Dubrovinskaia, M.Hanfland, A. I.Abrikosov, T.Bin Masood, D.Laniel, V. B.Prakapenka, A. F.Goncharov, M. I.Katsnelson, T.Fedotenko, A. V.Ponomareva, I. A.Abrikosov, L.Dubrovinsky, S.Chariton. High-pressure synthesis of Dirac materials: Layered van der Waals bonded BeN4 polymorph. Phys. Rev. Lett., 126, 175501(2021).
[37] V. B.Prakapenka, A. V.Ponomareva, S.Chariton, A. F.Goncharov, I. A.Abrikosov, E.Bykova, M. F.Mahmood, M.Bykov, L.Dubrovinsky. Stabilization of polynitrogen anions in tantalum–nitrogen compounds at high pressure. Angew. Chem., Int. Ed., 60, 9003-9008(2021).
[38] F.Tasnádi, I. A.Abrikosov, K.Glazyrin, M.Bykov, G.Aprilis, H.-P.Liermann, E.Koemets, A. V.Ponomareva, N.Dubrovinskaia, L.Dubrovinsky, J.Tidholm, T.Fedotenko, E.Bykova. High-pressure synthesis of a nitrogen-rich inclusion compound ReN8·
[39] A. V.Ponomareva, L.Dubrovinsky, M.Hanfland, S.Chariton, A. F.Goncharov, I. A.Abrikosov, P.Sedmak, M.Mahmood, M.Bykov, S.Khandarkhaeva, N.Dubrovinskaia, H. P.Liermann, J.Tidholm, E.Bykova, F.Tasnádi, T.Fedotenko, V.Prakapenka. High-pressure synthesis of metal–inorganic frameworks Hf4N20·N2, WN8·N2, and Os5N28·3N2 with polymeric nitrogen linkers. Angew. Chem., Int. Ed., 59, 10321-10326(2020).
[40] J.Yuan, H.Gao, H.-T.Wang, C.Liu, J.Sun, D.Xing, K.Xia. A novel superhard tungsten nitride predicted by machine-learning accelerated crystal structure search. Sci. Bull., 63, 817-824(2018).
[41] Y.Han, J.Sun, J.Wang, H.Gao. Enhancing crystal structure prediction by decomposition and evolution schemes based on graph theory. Fundam. Res., 1, 466-471(2021).
[42] Z.Guo, J.Wang, H.Gao, J.Sun. Determining dimensionalities and multiplicities of crystal nets. npj Comput. Mater., 6, 143(2020).
[43] J.Sun, C. J.Pickard, C.Liu, H.Gao, R. J.Needs, Y.Wang, H.-T.Wang, D.Xing. Multiple superionic states in helium–water compounds. Nat. Phys., 15, 1065-1070(2019).
[44] J.Sun, Q.Gu, D.Xing. Superconducting single-layer T-graphene and novel synthesis routes. Chin. Phys. Lett., 36, 097401(2019).
[45] M.Miao, H.-T.Wang, D.Xing, Y.Wang, A.Hermann, J.Sun, C. J.Pickard, C.Liu, R. J.Needs, H.Gao. Plastic and superionic helium ammonia compounds under high pressure and high temperature. Phys. Rev. X, 10, 021007(2020).
[46] A.Hermann, R. J.Needs, J.Sun, H.-T.Wang, C. J.Pickard, D.Xing, H.Gao, C.Liu. Coexistence of plastic and partially diffusive phases in a helium-methane compound. Natl. Sci. Rev., 7, 1540-1547(2020).
[47] J.Shi, H.Gao, C.Liu, J.Wang, Y.Han, J.Sun, D.Xing, X.Lu, H. T.Wang. Mixed coordination silica at megabar pressure. Phys. Rev. Lett., 126, 035701(2021).
[48] J.Yuan, J.Sun, J.Wang, C.Ding, H.Gao, Y.Han. High energy density polymeric nitrogen nanotubes inside carbon nanotubes. Chin. Phys. Lett., 39, 036101(2022).
[49] J.Furthmüller, G.Kresse. Efficient iterative schemes for
[50] P. E.Bl?chl. Projector augmented-wave method. Phys. Rev. B, 50, 17953-17979(1994).
[51] X.Zhou, G. I.Csonka, K.Burke, A.Ruzsinszky, G. E.Scuseria, O. A.Vydrov, J. P.Perdew, L. A.Constantin. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett., 100, 136406(2008).
[52] G.Kresse, D.Joubert. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B, 59, 1758-1775(1999).
[53] H.Krieg, S.Ehrlich, J.Antony, S.Grimme. A consistent and accurate
[54] L.Goerigk, S.Ehrlich, S.Grimme. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem., 32, 1456-1465(2011).
[55] I.Tanaka, A.Togo. First principles phonon calculations in materials science. Scr. Mater., 108, 1-5(2015).
[56] A. L.Tchougréeff, V. L.Deringer, R.Dronskowski, S.Maintz. Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids. J. Comput. Chem., 34, 2557-2567(2013).
[57] V. L.Deringer, S.Maintz, A. L.Tchougréeff, R.Dronskowski. LOBSTER: A tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem., 37, 1030-1035(2016).
[58] A.Otero-de-la-Roza, V.Lua?a, A. M.Pendás, M. A.Blanco. Critic: A new program for the topological analysis of solid-state electron densities. Comput. Phys. Commun., 180, 157-166(2009).
[59] E. R.Johnson, A.Otero-de-la-Roza, V.Lua?a. Critic2: A program for real-space analysis of quantum chemical interactions in solids. Comput. Phys. Commun., 185, 1007-1018(2014).
[60] K.Momma, F.Izumi.
[61] Y.Jean, J. K.Burdett, F.Volatron. An Introduction to Molecular Orbitals(1993).
[62] S.Limpijumnong, W. R. L.Lambrecht. Homogeneous strain deformation path for the wurtzite to rocksalt high-pressure phase transition in GaN. Phys. Rev. Lett., 86, 91-94(2001).
[63] F.Ali Sahraoui, N.Bouarissa, S.Zerroug. Ab initio calculations of yttrium nitride: Structural and electronic properties. Appl. Phys. A, 97, 345-350(2009).
[64] K.Bao, D.Duan, D.Li, S.Wei, T.Cui, F.Tian, Z.Liu, H.Yu, B.Liu, W.Wang. Bonding properties of aluminum nitride at high pressure. Inorg. Chem., 56, 7494-7500(2017).
[65] R. J.Needs, C. J.Pickard. High-pressure phases of nitrogen. Phys. Rev. Lett., 102, 125702(2009).
[66] I. C.Tranca, R. Y.Rohling, E. J. M.Hensen, E. A.Pidko. Correlations between density-based bond orders and orbital-based bond energies for chemical bonding analysis. J. Phys. Chem. C, 123, 2843-2854(2019).
[67] J.Contreras-García, S.Keinan, W.Yang, E. R.Johnson, P.Mori-Sánchez, A. J.Cohen. Revealing noncovalent interactions. J. Am. Chem. Soc., 132, 6498-6506(2010).
[68] K.Schulten, A.Dalke, W.Humphrey. VMD: Visual molecular dynamics. J. Mol. Graphics, 14, 33-38(1996).
[69] S. J.Jacobs, M. J.Kamlet. Chemistry of detonations. I. A simple method for calculating detonation properties of C–H–N–O explosives. J. Chem. Phys., 48, 23-35(1968).
[70] J.Zhang, A. R.Oganov, X.Li, H.Niu. Pressure-stabilized hafnium nitrides and their properties. Phys. Rev. B, 95, 020103(2017).

Set citation alerts for the article
Please enter your email address
© Copyright 2018-2021 | Chinese Laser Press. All Rights Reserved 沪ICP备15018463号-20