Dongdong Kang, Kai Luo, Keith Runge, S. B. Trickey. Two-temperature warm dense hydrogen as a test of quantum protons driven by orbital-free density functional theory electronic forces[J]. Matter and Radiation at Extremes, 2020, 5(6): 064403
Copy Citation Text
We consider a steady-state (but transient) situation in which a warm dense aggregate is a two-temperature system with equilibrium electrons at temperature Te, ions at Ti, and Te ≠ Ti. Such states are achievable by pump–probe experiments. For warm dense hydrogen in such a two-temperature situation, we investigate nuclear quantum effects (NQEs) on structure and thermodynamic properties, thereby delineating the limitations of ordinary ab initio molecular dynamics. We use path integral molecular dynamics (PIMD) simulations driven by orbital-free density functional theory (OFDFT) calculations with state-of-the-art noninteracting free-energy and exchange-correlation functionals for the explicit temperature dependence. We calibrate the OFDFT calculations against conventional (explicit orbitals) Kohn–Sham DFT. We find that when the ratio of the ionic thermal de Broglie wavelength to the mean interionic distance is larger than about 0.30, the ionic radial distribution function is meaningfully affected by the inclusion of NQEs. Moreover, NQEs induce a substantial increase in both the ionic and electronic pressures. This confirms the importance of NQEs for highly accurate equation-of-state data on highly driven hydrogen. For Te > 20 kK, increasing Te in the warm dense hydrogen has slight effects on the ionic radial distribution function and equation of state in the range of densities considered. In addition, we confirm that compared with thermostatted ring-polymer molecular dynamics, the primitive PIMD algorithm overestimates electronic pressures, a consequence of the overly localized ionic description from the primitive scheme.
I. INTRODUCTION
Laboratory advances have created new avenues of investigation into the warm dense matter (WDM) condensed-phase-state regime. Dynamic compression methods1–5 and high-power laser ablation6–8 allow the exploration of WDM over wide temperature–pressure ranges. Typical experimental creation of WDM deposits energy predominantly into either electrons or ions on time scales that are generally of the order of nanoseconds or shorter. In fact, for laser excitation, pulse durations can be in the femtosecond time domain. Concurrent heating and measurement of the electron subsystem temperature during 25 fs x-ray pulses have been reported.9 WDM produced by dynamic compression is usually in short-lived, spatially inhomogeneous states, and therefore detailed knowledge of such nonequilibrium (transient) WDM states is important.10–13
An interesting and important category is the two-temperature case: electrons at temperature Te and ions at Ti, with Te ≠ Ti. Over 40 years ago, Anisimov et al.14 proposed a two-temperature model for the interpretation of laser-pulse-excited electron emission from a metal surface. Hohlfeld et al.15 proposed three regimes to describe the response of a system to subpicosecond laser pulses. The first regime is characterized by ballistic transport of the excited electrons, which lasts until electron–electron scattering leads to a Te that, in the second regime, is not equal to Ti. In the third regime, ion–electron coupling results in an eventual thermal equilibrium state of the whole system, described by a single temperature, Te = Ti. Meanwhile, Murillo et al.16,17 predicted that heating the electrons creates an instantaneous response that leads to spontaneous ionic heating before thermal equilibration in the plasma regime.
A particularly pertinent description can be found in Ref. 18. Those authors considered “… ions at a temperature Ti much less than the measured electron temperature Te of 10 eV” treated by orbital-free density functional theory (OFDFT) driving ab initio molecular dynamics (AIMD)19–22 at “…different electronic and ionic temperatures. Such simulations correspond to separate equilibrium states of ions and electrons, but to an out-of-equilibrium state of the whole system. Since the electron system is treated in the simulations within density functional theory at Te, the ion–electron system is frozen in the out-of-equilibrium state of decoupled temperatures.” Other examples of the use of models with differing ion and electron temperatures to interpret the equation of state, optical, and transport properties of WDM include those in Refs. 23–27.
Here, we use the same two-temperature approach to treat hydrogen, but with far more advanced density functional approximations (see below). The majority abundance of hydrogen in the universe makes precise knowledge of its behavior in WDM state conditions crucial for modeling the interiors of giant planets.28 Its nuclear properties make hydrogen crucial for inertial confinement fusion experiments.29,30 A free-electron x-ray laser pump–probe measurement of dense cryogenic hydrogen is an important example.31–33 In those experiments, a pair of 300 fs, 92 eV pulses were separated by a time delay 0 < Δt ≤ 5 ps. Depending on the pump intensity (19 TW/cm2 or 27 TW/cm2), from roughly Δt = 1 ps onward to 5 ps, the electron temperature Te (from hydrodynamic analysis) was about 1.75 eV, while the ion temperature Ti was about 0.35–0.4 eV. The Spitzer equilibration time for the two temperatures to stabilize was calculated to be about 0.9 ps, a value consistent with observation. Several of the conclusions were confirmed by AIMD simulations driven by free-energy DFT.31,32
In AIMD, Born–Oppenheimer forces from DFT drive classical point ions. The ionic configuration space is then sampled with molecular dynamics. Applications to finite-T systems should use free-energy DFT calculations,34 but it is not unusual to see ground-state approximate functionals used with finite-T densities. Such was the case in Refs. 31 and 32. Although by now the approach has been applied extensively, there are three substantial challenges to such free-energy-DFT-based AIMD simulations that are highly relevant to understanding two-temperature hydrogen.
The most important of these three challenges concerns nuclear quantum effects (NQEs) on structure and thermodynamic properties.35 NQEs are particularly significant for low-mass elements such as hydrogen and its isotopes.36 An especially significant issue is the variation of NQEs as a system of low-mass ions traverses a wide range of state conditions. Several studies have been conducted using path integral techniques for the description of warm dense hydrogen or deuterium, with path integral molecular dynamics (PIMD) and PIMD-centroid approaches being frequent choices.37–46 As reported in the literature,37–39 NQEs have significant impacts on phase transitions between solid molecular hydrogen at megabar pressures, as well as the melting behavior of dense hydrogen.41,42 Furthermore, NQEs play an important role in the accurate description of the electronic structure of dense hydrogen. With the inclusion of the quantum nature of ions, the metallization, dissociation, and transport properties of hydrogen at high pressures exhibit strikingly different behaviors from what is found in conventional calculations with classical ions.36,43–48 Those differences cannot be described sufficiently even with the quasiharmonic approximation.37
Second, and more generically, the cost of standard Kohn–Sham (KS) DFT49 calculations scales with the cube of the number of occupied KS orbitals. As the temperature increases, the computational cost of conventional KS-DFT inexorably becomes prohibitive. A promising tool for material modeling at high temperatures is therefore the orbital-free form of KS theory (OFDFT), because of its linear scaling of computational cost with system size. An approximately linear scaling method that also does not depend on the system state to achieve sparsity is the “extended first-principles MD” scheme.50 This replaces high-lying, comparatively low-occupation KS states with plane waves, thereby sacrificing orthogonality but retaining atomic shell structure. It has been used to attempt to delineate the validity of OFDFT, but only in the context of the outmoded and fundamentally flawed Thomas–Fermi (TF) noninteracting free-energy functional and the ground-state local spin density approximation for the exchange-correlation (XC) free energy.51 By contrast, some state-of-the-art orbital-free noninteracting free-energy functionals have been developed recently and have been applied successfully to WDM.52–54 These substantially improve the achievable accuracy of OFDFT calculations for WDM and hence go well beyond the TF approximation that was used, for example, in Ref. 18.
Third, as remarked above, the majority of finite-T AIMD calculations have used ground-state XC functionals evaluated with finite-T densities. This “ground-state approximation” is now known to introduce nontrivial errors in both the equation of state and the principal Hugoniot.55–58 XC thermal effects need to be included in the WDM regime by use of genuine XC free-energy functionals.
The advance of OFDFT approximations for both noninteracting and XC free energies provides the opportunity to explore whether NQEs can be treated adequately by path integral simulations driven by OFDFT calculations. If successful, this would open new possibilities for large-scale detailed simulations of important WDM systems. Here, we investigate precisely that potential payoff. We study the structure and thermodynamic properties of two-temperature warm dense hydrogen using PIMD simulations for protons driven by electronic forces obtained from finite-temperature OFDFT. The effects of the OFDFT functional approximation are assessed by comparison with conventional KS-DFT calculations. With that comparison in hand, we then focus on the physics of the hydrogen problem by systematic assessment of NQEs. This is done by comparisons between orbital-free Born–Oppenheimer molecular dynamics (which for brevity we denote as OFMD) simulations and orbital-free-driven path integral PIMD (PI-OFMD) simulations. To avoid the well-known and much-studied complications of the molecular H2 to atomic H liquid–liquid transition, we consider ion temperatures and densities that, from the best available evidence,41 should, for the most part, correspond to the atomic liquid. Details are given below.
In this paper, we first briefly describe the PIMD methods for protons and finite-temperature OFDFT methods for electrons in Sec. II. Then, in Sec. III, we describe the computational details for two-temperature warm dense hydrogen. The results and a discussion are presented in Sec. IV in which the calibrating comparisons with other methods, NQEs on radial distribution functions, and equation of state are discussed in detail. Finally, we give a brief conclusion in Sec. V.
II. METHODS
A. Path integral molecular dynamics for protons
This subsection provides a brief description of the PIMD treatment for protons. Note that the temperatures under consideration in this work are higher than the quantum degeneracy temperature,59 and hence proton exchange effects are neglected in the following formulation. The canonical partition function of N identical ions of mass m at inverse temperature β = 1/kBTi (kB is the Boltzmann constant) takes the well-known discretized path integral form60,61where is the number of discretized points, , and is the potential energy of the system at the imaginary time slice s for system configuration {ri}. For the two-temperature system, is conventionally noted as the electronic free energy, although the interionic potential is included.
In Eq. (1), the canonical partition function of a quantum system has been expressed as a configurational integral of Boltzmann-weighted continuous paths. Those closed paths are discretized through the Trotter approximation62 into “beads” circularly connected via harmonic springs, Thus, as is well known, an N-particle quantum system can be made approximately isomorphic to a classical system consisting of N ring polymers, in which each quantum particle is mapped onto a closed, flexible polymer of beads.63,64 The approximation becomes a true isomorphism in the limit , i.e., .
To sample the configuration space of the classical polymer system using MD simulations, and thereby obtain the thermodynamic properties of the isomorphic quantum system, an effective Hamiltonian is defined bywhere m′ is the fictitious mass that determines the efficiency of sampling of the polymer configurations, is the fictitious momentum conjugate to the position , and the effective potential isThe equations of motion then follow:
The foregoing formulation comprises the primitive PIMD algorithm. Because the harmonic confinement coupling grows as , this algorithm is known to have practical limitations with respect to configuration space sampling.65 Craig and Manolopoulos66 extended the algorithm to ring-polymer molecular dynamics (RPMD) for simulating the fictitious quantum dynamics approximately. In RPMD, the fictitious bead mass m′ is chosen to be the physical mass m, and the harmonic bead-coupling and potential-energy terms are taken to be times larger than their counterparts in Eq. (4). It can be shown that these choices do not have any impact on the generation of thermodynamic equilibrium average quantities.67 Thus, we adopt thermostatted RPMD (TRPMD)68 with normal mode transformation69 for the bead coordinates to perform the PIMD simulations. At the end, we give a brief comparison of results from the primitive PIMD algorithm.
The ensemble averages for thermodynamic quantities can be derived from the path integral representation of the partition function in Eq. (1). In particular, the thermodynamic estimator for the total free energy is contributed by the quantum kinetic energy estimator61the potential-energy (electronic free-energy) estimatorand the entropy contribution from the nuclear configurations. Here, we neglect the difference in the ionic configuration entropy between the classical and quantum treatment of ions.
The thermodynamic estimator for the pressure p iswhere V is the system volume. The first and second terms are the contributions of the ionic quantum motions to the total pressure, while the third term is the electronic contribution (in some of the literature, the electron pressure is known as the excess pressure). Averaging these estimators along the MD trajectory yields the corresponding observable thermodynamic quantities.
B. Finite-temperature OFDFT for electrons
In ab initio PIMD, the interpolymeric interaction at the imaginary-time slice s has two parts, namely, the ion–ion electrostatic interaction and the electron–ion interaction energy. The latter contribution can be calculated via free-energy DFT.34 In this, the electronic free energy is obtained by minimizing the grand canonical potential with respect to the electron density n(r, Te). The grand canonical potential has the form52where v(r) is the external potential on the electrons corresponding to electron density n and μ is the chemical potential. The free-energy functional F[n] is composed of the noninteracting free energy Fs[n], the classical Coulomb repulsion energy (i.e., Hartree energy) FH[n], and the XC free energy Fxc[n],The Hartree energy has the same form as in zero-temperature DFT,The noninteracting free energy iswhere Ts[n] and Ss[n] are the noninteracting kinetic free energy and entropy, respectively. The XC free energy is the excess electron–electron interaction energy with respect to the Hartree energy plus the differences in the kinetic free energy and entropy between the fully interacting system and the noninteracting reference system, to wit,where Uee[n] is the full electron–electron Coulomb interaction free energy.
In conventional KS-DFT, a sophisticated scheme exploits the one-electron orbitals of the noninteracting system to construct the electron density of the real system and thereby the total free energy. The advantage of conventional KS-DFT is that the noninteracting free-energy functionals Ts[n] and TeSs[n] can be constructed exactly from the one-electron orbitals and their Fermi–Dirac occupations, thereby giving an explicit Euler equation once a suitable approximate Fxc is provided.
In contrast to conventional KS-DFT, in OFDFT, the noninteracting functionals Ts[n] and Ss[n] are formulated directly in terms of the electron density rather than the KS orbitals. Doing so requires approximations. Given these, minimization of the grand canonical potential in Eq. (8) with respect to the electron density n(r, Te) gives the Euler–Lagrange equationThe computational cost of solving this equation scales linearly with system size and is essentially temperature-independent. The challenge is that both accurate noninteracting and XC free-energy functionals are essential ingredients for successful application of OFDFT to WDM.
Recently, a framework for generating constraint-based nonempirical orbital-free generalized gradient approximations for the noninteracting free-energy functional has been explored,52 and two distinct noninteracting free-energy functionals, VT84F53 and LKTF,54,70 have been developed. More or less concurrently, several explicitly temperature-dependent XC functionals have also been proposed at both the local density approximation (LDA) level of refinement71,72 and the generalized gradient approximation (GGA) level.73 They have been shown to have significant thermal effects on equations of state, principal Hugoniots, and optical properties of WDM.56,57 The opportunity opened by these functionals that is explored here is to broaden both the scope and the accuracy of ab initio PIMD calculations of NQEs.
III. COMPUTATIONAL DETAILS
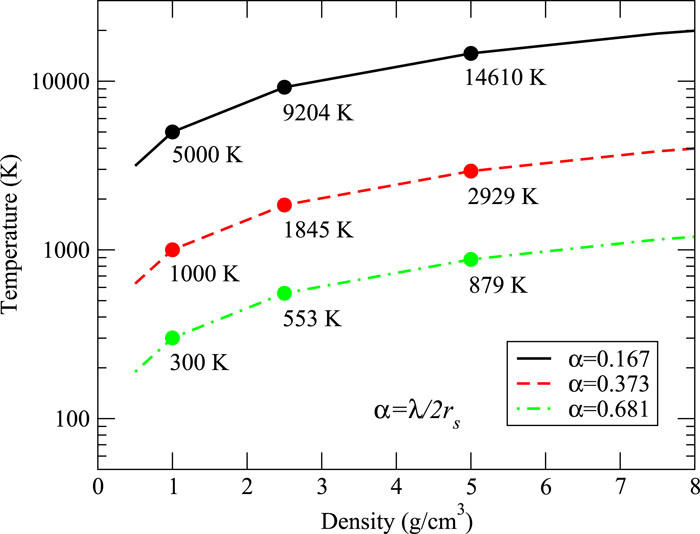
To investigate the impact of NQEs on the material properties of a sensitive two-temperature system, we have performed extensive PI-OFMD simulations of two-temperature warm dense hydrogen. The electron temperatures studied were Te = 20 kK, 50 kK, and 100 kK. The densities and ion temperatures were specified in terms of a dimensionless scale parameter α ≔ λ/2rs based on the ionic thermal de Broglie wavelength and the ionic Wigner–Seitz radius rs = (3V/4πN)1/3, where h is the Planck constant, m is the ionic mass, Ti is the ion temperature, and V is the system volume. The parameter α is the ratio of the effective quantum size of the ions to their mean separation, and thus it provides a measure of the degree of ionic quantum nature. The densities, ion temperatures, and α values used are displayed in Fig. 1. For the density of 1 g/cm3, we chose Ti = 300 K, 1000 K, and 5000 K, corresponding to α = 0.681, 0.373, and 0.167, respectively. At 2.5 g/cm3, we used Ti = 553 K, 1845 K, and 9204 K, respectively, and at 5 g/cm3, 879 K, 2929 K, and 14 610 K, respectively.
Figure 1.The state points of the density and ion temperature used in the present simulations. The parameter α takes values of 0.167, 0.373, and 0.681, corresponding to the three curves from top to bottom, respectively. The densities of these state points are 1 g/cm3, 2.5 g/cm3, and 5 g/cm3, respectively.
The phase diagram of the real physical system is, of course, at local thermodynamical equilibrium, Ti=Te. This phase diagram therefore provides only limited context for these α choices. At Ti = 300 K and the lower densities chosen, the equation of state that we find for the nonequilibrium (two-temperature) system gives pressures for which the equilibrium physical system is predicted to be in a molecular liquid state41,43 or a molecular solid.74 This distinction from the results of the equilibrium calculations does not persist at the higher temperatures.
The PIMD simulations used the i-PI code75 combined with a locally modified version of the PROFESS package76,77 for OFDFT calculations. In terms of Eq. (4), the discretized Trotter beads run on the potential surface generated by the harmonic interaction between the neighboring beads and the interpolymeric interaction calculated by OFDFT. The accurately parametrized finite-temperature local density XC free-energy functional KSDT71 was used. (Use of the corrKSDT LDA functional would not alter the results for the state conditions studied.78) The nonempirical constraint-based generalized-gradient-approximation (GGA) noninteracting free-energy functional LKTF54 was used. This is the finite-temperature extension of the noninteracting kinetic energy functional LKT.70 LKT, in turn, is specifically designed to meet rigorous constraints when used with pseudopotentials, as is the case here. For comparative purposes, we also used VT84F,53 an earlier nonempirical constraint-based noninteracting free-energy approximation.
We used a local pseudopotential (LPP) for all of the OFDFT calculations. Details regarding this can be found in Ref. 52. For comparison, conventional Kohn–Sham AIMD (KSMD) calculations were done with that same LPP unless otherwise noted. In the OFDFT calculations with PROFESS, the numerical grid for real-space integrations was set to 64 × 64 × 64 to ensure free-energy and pressure convergence.
Periodic supercells including 128–250 atoms were employed, depending on the bulk density, and the body-centered cubic configuration was used as the starting ionic structure for all the MD simulations, both conventional KSMD and OFMD, in this study.
In PIMD, the Trotter bead number was . AIMD with classical ions but with the same algorithmic and code structure was achieved by setting . The stochastic path integral Langevin equation thermostat was used to improve the sampling efficiency. See Ref. 79 for details. After equilibration of 5000 MD steps, 15 000 configurations with time steps of 0.2 fs–0.3 fs were generated for taking the thermodynamical averages.
For comparison with conventional KSMD, we performed simulations using the Quantum-ESPRESSO package.80 To achieve consistency of comparison with the orbital-free calculations, the KSDT XC free-energy functional was used. The projector augmented wave procedure was employed with a 120 Ry plane-wave energy cutoff. A sufficient number of energy bands were considered in order to make the highest occupied band energy higher than the chemical potential by at least 20kBTe. Γ-point Brillouin-zone sampling was used.
IV. RESULTS AND DISCUSSION
A. Comparison with other methods
Insight into the accuracy of the noninteracting free-energy functionals is provided by two preliminary studies. First, we carried out KSMD and OFMD calculations (no NQEs) with the same LPP in local thermodynamic equilibrium at fixed, comparatively low temperature, Te = Ti = 2 kK for three densities, rs = 1.05, 1.10, and 1.25. The results for the modern LKTF54,70 and VT84F53 GGA noninteracting functionals and finite-temperature Thomas–Fermi functional (TTF)52 are shown in Table I. It can be seen that LKTF is somewhat better in terms of isothermal pressure error than VT84F or TTF, in the sense that the range of LKTF fractional error is smallest of the three for the density range considered. It is also important to note that the TTF error is deceptive in that TTF does not give bound crystals in the static lattice limit and has other failures even for hydrogen.53
rs
P (KSMD)
P (LKTF)
P (VT84F)
P (TTF)
1.05
1558.7
1453.0(0.068)
1401.7(0.101)
1636.1(−0.049)
1.10
1146.9
1061.0(0.075)
1012.2(0.117)
1224.3(−0.067)
1.25
470.9
416.8(0.115)
377.3(0.199)
536.4(−0.139)
Table 1. Comparison of conventional KSMD and OFMD electronic pressures (GPa) at equilibrium, Te = Ti = 2 kK. The fractional error for OFMD with respect to the KSMD pressure is shown in parentheses. The KSDT XC free-energy functional was used in all cases.
Next, we investigated the behavior of the three noninteracting functionals (LKTF, VT84F, and TTF) for the two-temperature situation, again with OFMD calculations compared with KSMD calculations (no NQEs), all using the same LPP. The two-temperature warm dense hydrogen had electron temperature Te = 20 kK and bulk density ρ = 1 g/cm3, with 300 kK ≤ Ti ≤ 20 kK. Comparisons of the electronic pressure are shown in Fig. 2. The gross failure of TF is now obvious, as is the improvement of LKTF compared with VT84F. However, as in the preceding case, LKTF again underestimates the electronic pressure relative to the KSMD values. As before, the underestimate is smooth and weakly varying. It decreases slowly and evenly with increasing ion temperature, from about 19% at Ti = 300 K to about 11% at Ti = 20 kK.
Figure 2.Comparisons of the electronic pressure calculated from OFMD simulations with the different noninteracting free-energy functionals (namely, TTF,52 VT84F,53 and LKTF54,70) vs KSMD simulations, all with the same LPP. Te = 20 kK and ρ = 1 g/cm3.
On the basis of these two comparisons, we make the quite plausible assumption that this smooth offset is inconsequential for reasonable estimates of NQEs. Therefore, for the remainder of this study, we use LKTF for the OFMD and PI-OFMD calculations.
Next, we compare the PI-OFMD quantum nuclear corrections to the hydrogen total free energy and pressure with corresponding results from coupled electron–ion Monte Carlo (CEIMC) calculations81 in the case of thermodynamic equilibrium states. Specifically, we considered Ti = Te = 2000 K and three densities corresponding to ionic Wigner–Seitz radii rs = 1.05 bohrs, 1.10 bohrs, and 1.25 bohrs. (Note that since the system is hydrogen, the ionic and electronic Wigner–Seitz radii are identical.) As shown in Table II, the quantum nuclear corrections from PI-OFMD are substantially larger than those from CEIMC for both the total free energy and pressure.
rs
α
ΔF (mhartree/atom)
ΔP (GPa)
ΔP/P (%)
CEIMCa
1.05
0.350
4.0(7)
7(3)
0.4
1.10
0.334
3.8(3)
9(1)
0.7
1.25
0.294
2.8(5)
5(1)
1.0
PI-OFMD
1.05
0.350
7.0(7)
27.7(4)
1.9
1.10
0.334
6.3(7)
23.6(3)
2.2
1.25
0.294
4.1(6)
11.1(2)
2.5
Table 2. NQE corrections to the total free energy and pressure of hydrogen from CEIMC and PI-OFMD calculations at an ion temperature Ti = 2000 K. In PI-OFMD, Te = 2000 K, while in CEIMC, the NQE corrections are obtained based on the zero-temperature potential-energy surface.81 The total energy includes the ionic kinetic energy and electronic free energy, Ftot = ϵkin + ϵpot, as defined by Eqs. (5) and (6), respectively. Corrections are defined as ΔF = (Ftot − Ftot,classical)/N and ΔP = P − Pclassical, respectively. The ratio of the pressure corrections to the pressure obtained classically, ΔP/P, is presented. Statistical errors are reported in parentheses as the uncertainty in the last digit.
In the CEIMC calculation,81 in order to assess NQEs on the thermodynamic properties, path integral Monte Carlo was performed for the protons on the potential-energy surface defined by the zero-temperature reptation quantum Monte Carlo method. In this work, the quantum protons are driven by the on-the-fly potential obtained from OFDFT calculations of electrons at nonzero temperatures. Different treatments of electronic structure lead to the much lower NQE estimates from CEIMC. It is important to note, however, that both sets of NQE corrections (to both the free energy and pressure) increase with density; see Table II. This accords with basic expectations about NQEs (specifically, a sort of excluded-volume effect) and reinforces the conclusion that the two schemes are describing how the same nuclear effect is manifested in very different physical circumstances. See below for further discussion regarding that effect in the equation of state.
B. Radial distribution function
To assist in understanding NQEs on the structure of two-temperature hydrogen, we calculated the radial distribution functions (RDFs) from PI-OFMD and compared these with their classical counterpart OFMD results. In PIMD, the RDF can be calculated viawhere ni(r) is the mean number of atoms in a shell of width Δr at distance r, ni,0 is the mean atom density, and 〈〉PI is the PI ensemble average.
Figure 3 shows the RDF of two-temperature hydrogen at the ion temperature and density state points considered in this study when Te = 20 kK. For the three cases that correspond to α = 0.167, the RDFs from OFMD and PI-OFMD calculations completely overlap one another. When the parameter α is increased to 0.373, the first RDF peak is broadened by the inclusion of NQEs. The degree of broadening is similar in the three cases.
Figure 3.Comparisons of the radial distribution function from OFMD and PI-OFMD for two-temperature hydrogen at different ion temperatures and densities. The parameter α is 0.167, 0.373, and 0.681 from the top to bottom panels. The electron temperature is 20 kK.
When α is raised to 0.681, however, the RDFs from PI-OFMD calculations exhibit distinctly different behaviors from those from OFMD calculations. The structure delivered by the OFMD is the cubic solid from which the simulation started, with a well-defined rather narrow first peak, followed by a pair of structured peaks for all three densities. For all three densities, the PI-OFMD results lower the first peak and broaden it slightly, while softening and smoothing the subsequent pair of peaks into a somewhat structured single peak. At the highest density, a third peak is pushed into the picture, again modestly broadened and lowered by NQEs relative to the OFMD result. The RDF behavior confirms that in quantum simulations, the protons vibrate in a larger volume around their equilibrium positions than in the classical case. This enhances the anharmonic vibrational effects, thereby lowering the melting temperature.42,82 Since the RDF is directly related (by Fourier transformation) to the structure factor, which is a key quantity in interpreting x-ray scattering measurements, NQEs on RDF will inevitably affect the x-ray scattering properties of light-atom matter such as hydrogen under high pressures and at low temperatures.40
That said, there is a question: Why does the atomic solid appear in the OFMD calculations for α = 0.681? Someone unfamiliar with the LPP utilized here might speculate that it is implicated. However, it was shown in Ref. 52 that it is realistic under a rather large range of state conditions. To the extent that one trusts a hard (small-core) but ground-state PAW for extreme state conditions, the validity of the LPP has also been confirmed by comparison of KSMD using LPP results with results from KSMD using PAW; see Refs. 52 and 53. The occurrence of the atomic solid is thus diagnosed as arising from the orbital-free approximate noninteracting free-energy functional. This diagnosis is confirmed by direct comparison of KSMD and OFMD calculations with the same LPP. We used Te = 20 kK for ρ = 1 g/cm3, Ti = 300 K, and for ρ = 5 g/cm3, Ti = 879 K, i.e., α = 0.681. We also carried out KSMD calculations with standard Quantum-Espresso PAW data sets for the same conditions. Figure 4 shows clearly that the atomic solid is a consequence of the orbital-free approximate functional. The KSMD RDFs (which are identical for LPP or PAW on the scale of the figure) are obviously liquid in character, while the OFMD RDFs show the characteristic solid peak sequence.
Figure 4.Comparisons of RDFs of two-temperature hydrogen obtained from KSMD (LPP and PAW) and OFMD calculations at the state points (5 g/cm3, 879 K) and (1 g/cm3, 300 K), corresponding to dimensionless size α = 0.681, for Te = 20 kK.
Despite these detailed differences, the RDFs provide an unmistakable semiquantitative criterion for the onset of meaningful NQEs. In Fig. 5, we display the ratio of the first peak height from OFMD (classical nuclei) to that for PI-OFMD as a function of the parameter α. What is qualitatively and at least roughly quantitatively indicated in this plot is that α ≈ 0.30 is a lower bound for meaningful NQEs. Independent calculations by some of us47 on the insulator–metal transition in hydrogen and deuterium corroborate this interpretation. In those other calculations, PIMD driven by accurate conventional KS-DFT calculations and by OFDFT also exhibit notable NQEs above α ≈ 0.30. Moreover, because of the ionic-mass dependence of α (smaller values for heavier ions means smaller NQEs), we suggest that α ≈ 0.30 as a semiquantitative lower bound for meaningful NQEs is universal for systems irrespective of different ionic compositions, not just for hydrogen. Obviously, this requires further investigation. The accuracy limitations of the LKTF functional used here do not alter these deductions about a roughly universal α.
Figure 5.Ratio of the first maximum in the RDF for the quantum and classical cases as a function of α.
We can examine the differing electron temperature effects on ionic structural properties by comparing the proton RDFs at the three electron temperatures Te = 20 kK, 50 kK, and 100 kK. Since the RDFs corresponding to the parameter α = 0.167 are indistinguishable, we present in Figs. 6 and 7 comparisons of the RDFs only for α = 0.373 and 0.681, respectively. For α = 0.373 at the higher density, 5 g/cm3, there are only slight RDF changes as Te is increased from 20 kK to 100 kK. The main effect is from NQEs, which, irrespective of Te, lower and broaden the first RDF peak modestly. At 1 g/cm3, the height of the first peak increases just enough to be perceptible as Te grows to 100 kK but still the stronger influence is that of NQEs. For α = 0.681 (Fig. 7), the outcome is similar but more pronounced. Only slight changes in the RDF appear over the entire Te range, whereas the NQEs are strong enough to wash out the crystalline order that OFMD produces. In short, because even the smallest out-of-equilibrium temperature ratio is very large, Te/Ti ≥ 6.8, the calculated RDFs are insensitive to growth in that out-of-equilibrium ratio.
Figure 6.Comparisons of the RDFs of two-temperature hydrogen at two state points (5 g/cm3, 2929 K) and (1 g/cm3, 1000 K), corresponding to dimensionless size α = 0.373, for Te = 20 kK, 50 kK, and 100 kK. Both OFMD and PI-OFMD results are shown.
To obtain further insight into NQEs, it is helpful to consider the spatial spread of the discretized path. A useful measure is the radius of gyration of the ith polymer. In PIMD, this is defined aswhere is the centroid of the ith polymer. Figure 8 shows changes in the radius-of-gyration distribution of the two-temperature hydrogen at Te = 20 kK. Given a fixed density, the mean radius of gyration increases as the ion temperature is decreased, with great broadening at the lowest temperatures. This distinct temperature effect clearly shows the nature of quantum delocalization of protons; i.e., the lower temperature results in a larger quantum “size” and stronger relative quantum fluctuation of particles. This behavior is also the reason why the RDFs from quantum simulations are quite different from the classical RDFs.
Figure 8.Distribution of the radius of gyration of the two-temperature hydrogen at Te = 20 kK and (from top to bottom) densities 1 g/cm3, 2.5 g/cm3, and 5 g/cm3.
The ionic, electronic, and total pressures calculated from OFMD and PI-OFMD simulations are summarized in Table III. The most obvious outcome is that the ionic pressure from quantum ions (PI-OFMD calculations) is significantly larger than from classical ions (OFMD calculations). The disparity ΔPi/Pi grows as the ratio of quantum proton size to the mean interproton distance, i.e., the parameter α, is increased. This unsurprising behavior is a consequence of the inclusion in the PI-OFMD simulations of the quantum kinetic energy contribution to the ionic pressure (via the harmonic interactions between neighboring beads in the classical isomorphic polymer).
Te (kK)
α
ρ (g/cm3)
Ti (K)
(GPa)
Pe (GPa)
ΔPe/Pe (%)
(GPa)
Pi (GPa)
ΔPi/Pi (%)
Pclassical (GPa)
P (GPa)
ΔP/P (%)
20
0.167
1.0
5 000
198.99
199.15
0.08
40.68
40.43
−0.60
239.67
239.58
−0.04
20
0.167
2.5
9 204
1815.79
1819.70
0.21
194.40
200.18
2.97
2010.20
2019.88
0.48
20
0.167
5.0
14 610
7389.67
7408.13
0.25
589.51
626.58
6.29
7979.17
8034.71
0.70
20
0.373
1.0
1 000
174.56
178.68
2.36
8.31
12.15
46.21
182.86
190.83
4.36
20
0.373
2.5
1 845
1725.10
1740.63
0.90
38.77
56.40
45.47
1763.87
1797.03
1.88
20
0.373
5.0
2 929
7151.90
7196.04
0.62
117.03
174.99
49.53
7268.92
7371.03
1.40
20
0.681
1.0
300
164.42
172.17
4.71
2.46
9.08
269.10
166.88
181.25
8.61
20
0.681
2.5
553
1689.21
1717.70
1.69
11.46
42.49
270.77
1700.67
1760.19
3.50
20
0.681
5.0
879
7060.90
7135.82
1.06
36.30
129.85
257.71
7097.19
7265.67
2.37
50
0.167
1.0
5 000
258.40
259.23
0.32
41.51
42.52
2.43
299.91
301.75
0.61
50
0.167
2.5
9 204
1921.09
1924.74
0.19
184.84
188.11
1.77
2105.93
2112.84
0.33
50
0.167
5.0
14 610
7557.53
7572.51
0.20
598.92
618.34
3.24
8156.45
8190.84
0.42
50
0.373
1.0
1 000
235.36
239.65
1.82
8.06
12.33
52.98
243.42
251.98
3.52
50
0.373
2.5
1 845
1836.57
1851.04
0.79
38.59
55.28
43.25
1875.16
1906.33
1.66
50
0.373
5.0
2 929
7322.82
7362.95
0.55
119.67
172.68
44.30
7442.49
7535.63
1.25
50
0.681
1.0
300
226.57
233.68
3.14
2.42
9.26
282.64
229.00
242.94
6.09
50
0.681
2.5
553
1801.53
1829.30
1.54
11.22
42.39
277.81
1812.76
1871.69
3.25
50
0.681
5.0
879
7230.73
7304.81
1.02
36.56
130.00
255.58
7267.29
7434.81
2.31
100
0.167
1.0
5 000
466.02
466.47
0.10
41.08
41.89
1.97
507.10
508.36
0.25
100
0.167
2.5
9 204
2248.03
2248.48
0.02
189.55
187.94
−0.85
2437.58
2436.41
−0.05
100
0.167
5.0
14 610
8027.68
8041.09
0.17
593.45
618.83
4.28
8621.13
8659.92
0.45
100
0.373
1.0
1 000
448.47
451.61
0.70
8.41
12.66
50.54
456.88
464.27
1.62
100
0.373
2.5
1 845
2166.42
2181.17
0.68
37.46
55.76
48.85
2203.88
2236.93
1.50
100
0.373
5.0
2 929
7797.50
7839.59
0.54
118.42
175.35
48.07
7915.92
8014.94
1.25
100
0.681
1.0
300
441.51
447.02
1.25
2.41
9.76
304.98
443.92
456.78
2.90
100
0.681
2.5
553
2135.69
2160.99
1.18
11.20
42.68
281.07
2146.89
2203.67
2.64
100
0.681
5.0
879
7708.87
7781.02
0.94
36.15
130.24
260.28
7745.02
7911.26
2.15
Table 3. Electronic, ionic, and total pressures of two-temperature hydrogen for various densities and electronic and ionic temperatures (Te, Ti) from OFMD and PI-OFMD calculations. Pe, Pi, and P are the electronic pressure, ionic pressure, and total pressure from PI-OFMD calculations respectively. , , and Pclassical are their OFMD counterparts with classical ions. The noninteracting free-energy functional LKTF is used. The dependence on the dimensionless size parameter α is also shown.
Observe that for a fixed α value, ΔPi/Pi stays nearly constant with respect to Te, with only small variations. ΔPi/Pi thus depends on the degree of quantum delocalization of protons rather than on the specific ion temperature and density conditions. The only exception is the case of small dimensionless size, α = 0.167. Even for this, variation of Te has almost no impact on ionic pressures. These behaviors are a consequence of the definition of as the ideal gas pressure for temperature equal to the MD-run average of the instantaneous thermostatted ionic temperature. For diverse technical reasons, that average temperature may differ slightly from the nominal (specified) ionic temperature. As a result, the ideal gas pressures calculated with the average and the nominal ionic temperatures may differ slightly. Close values of the average and nominal Ti, or, equivalently, close values of calculated with those two temperatures, indicate the thermostat quality. In our calculations, the difference between calculated from MD directly and the ideal gas pressure calculated with the nominal Ti does not exceed 2%. We note that for α = 0.167, there are a few anomalous cases with negative pressure increments. Those result from statistical fluctuations related to the closeness of ionic pressures obtained from OFMD and PI-OFMD simulations.
More interestingly, the electronic pressures from quantum simulations are larger than those from classical treatments of the protons. This increase of Pe by NQEs is below 5% at the temperature and density conditions considered here. This behavior should be attributed to the alteration of the electron density for quantum simulations of protons relative to their classical treatment. We note that the ionic configuration is significantly affected by NQEs, as shown in Fig. 3. This corresponds to a different external potential for the electrons to which the electron density must adapt. Inclusion of NQEs gives the protons a larger effective size than in the classical case, so the effective volume available for electrons is reduced and the electronic pressure goes up.
While the ionic pressure is dramatically raised by NQEs, the increment of total pressure with quantum protons is below 10% at the temperature and density conditions in this study. Nevertheless, the quantum nuclear corrections to the total pressure of two-temperature warm dense hydrogen are not negligible, and hence they should be considered when highly accurate equation-of-state data are required, especially for low-ion-temperature and high-density conditions.
The quantum nuclear corrections to the free energy are shown in Fig. 9. First, note that, similar to the pressure corrections, the differing electron temperature has almost no impact on free-energy corrections. More importantly, the free-energy corrections for protons increase approximately linearly with increasing density for all three cases, α = 0.167, 0.373, and 0.681. Therefore, the corrections to the free energy can be estimated in the entire nonequilibrium process through extrapolations from those somewhat limited PIMD simulations.
Figure 9.Quantum nuclear corrections to the free energy per hydrogen atom at the state points of ion temperature and density used. Results at Te = 20 kK, 50 kK, and 100 kK are presented for comparison.
As remarked at the outset, in this study, we used the TRPMD68 method to sample the ionic configuration space of the isomorphic polymer system rather than the primitive algorithm.61 For clarity about methodological effects, in Table IV, we show the difference between the electronic pressures obtained from the primitive algorithm and from TRPMD for the case ρ = 1 g/cm3. For the simulations with the number of beads , both algorithms yield almost the same results. But with the number of beads , we find that the electronic pressure is overestimated significantly by the primitive algorithm as compared with TRPMD results. This is consistent with the known sampling adequacy of ionic configurations for the primitive algorithm.67,83 The TRPMD technique can provide a solution to this problem with rapid, proper canonical sampling. This is simply a reminder of the effects of the choice of algorithm for the study of NQEs on equation-of-state or other thermodynamic properties of WDM.
Te (kK)
Ti (K)
(GPa)
Pe (GPa)
ΔP/P (%)
Primitive
20
300
136.8(1.0)
166.5(3.1)
21.7
20
1000
146.4(1.3)
167.0(2.0)
14.1
20
5000
169.5(1.0)
184.0(3.2)
8.6
TRPMD
20
300
137.6(0.2)
144.6(0.3)
5.1
20
1000
146.3(0.5)
150.4(0.7)
2.8
20
5000
168.2(2.0)
169.2(2.1)
0.6
Table 4. Comparisons of the electronic pressure Pe obtained from the primitive PIMD algorithm61 and TRPMD68 for hydrogen at ρ = 1 g/cm3. The results of simulations with classical protons are also presented by setting the bead number . Here, the noninteracting free-energy functional VT84F53 and the finite-temperature XC functional KSDT71 are used. The standard deviation is shown in parentheses.
In summary, we have performed PI-OFMD simulations for two-temperature warm dense hydrogen and have systematically estimated nuclear quantum effects upon its structure and thermodynamic properties. The use of OFDFT with PIMD has provided a less computationally intensive option than conventional KSMD for studying two-temperature effects. The best OFDFT noninteracting free-energy functional currently available (LKTF) combined with the LDA finite-temperature XC functional improves upon the ground-state approximation by the inclusion of an explicit T dependence. The combination gives electronic pressures that reasonably reproduce the trends in the KSDFT calculations but are offset. However, the offset is substantially smaller than with other noninteracting free-energy functionals, especially TF, although further improvement in Fs[n] is still needed.
We have identified an unexpected limitation of LKTF, namely, the appearance of spurious solid-like character in the RDF at the largest α = 0.681. Exploration of the cause is a consideration that needs to be taken into account in the development of a better Fs[n] approximation.
NQEs calculated with this methodology are substantially larger than those from the CEIMC methodology. We have suggested the very different (reversed) two-temperature relationships in the two methods as the most obvious plausible source of the discrepancy. The use in CEIMC of clamped nuclei to generate the Te = 0 electronic potential surface would in principle amplify the difference. That proposed identification is a matter for further methodological exploration. What is learned unequivocally from the two very different sets of circumstances and methods is the ubiquitous importance of NQEs for the equation of state.
Inclusion of ionic quantum effects alters the ionic RDFs perceptibly when the parameter α (defined as the ratio of the ionic thermal de Broglie wavelength to the mean interionic distance) is as large or larger than about 0.30. This interpretation is confirmed by examination of first-peak heights in the RDFs.
A significant physical finding is that variation of Te over a large range has only small to negligible effects on the ionic RDFs in the range of densities considered. This may be an indication of non-Born–Oppenheimer effects. Another possible reason is that the electronic structure does not change much at Te lower than the Fermi temperature. This is another issue that needs further investigation.
Importantly, NQEs raise both the ionic and electronic pressures. This is because quantum protons have a larger effective size than classical point ions. When highly accurate equation-of-state data for warm dense hydrogen are required, NQEs should not be ignored, especially under conditions of relatively low ionic temperature and high density. The extent to which this is true of other light elements needs to be ascertained on a case-by-case basis.
In addition, we have found that the use of the primitive algorithm of PIMD leads to an overestimation of electronic pressures of two-temperature warm dense hydrogen. Any attempt to use this primitive algorithm as a computational economy is therefore ill-advised.
[14] B. L. Kapeliovich, S. I. Anisimov, T. L. Perel’man. Electron emission from metal sufraces exposed to ultrashort laser pulses. Sov. Phys. JETP, 39, 375(1974).
[20] J. Hutter, D. Marx, J. Grotendorst. Ab initio molecular dynamics: Theory and implementation. Modern Methods and Algorithms of Quantum Chemistry, 301ff(2000).
[65] J. Grotendorst, M. E. Tuckerman, D. Marx, A. Muramatsu. Path integration via molecular dynamics. Quantum Simulations of Complex Many-Body Systems: From Theory to Algorithms, 269-298(2002).
Dongdong Kang, Kai Luo, Keith Runge, S. B. Trickey. Two-temperature warm dense hydrogen as a test of quantum protons driven by orbital-free density functional theory electronic forces[J]. Matter and Radiation at Extremes, 2020, 5(6): 064403