In quantum mechanics, when an electron is quickly ripped off from a molecule, a superposition of new eigenstates of the cation creates an electron wave packet that governs the charge flow inside, which has been called charge migration (CM). Experimentally, extracting such dynamics at its natural (attosecond) timescale is quite difficult. We report the first such experiment in a linear carbon-chain molecule, butadiyne (C4H2), via high-harmonic spectroscopy (HHS). By employing advanced theoretical and computational tools, we showed that the wave packet and the CM of a single molecule are reconstructed from the harmonic spectra for each fixed-in-space angle of the molecule. For this one-dimensional molecule, we calculate the center of charge ⟨ x ⟩ ( t ) to obtain vcm, to quantify the migration speed and how it depends on the orientation angle. The findings also uncover how the electron dynamics at the first few tens to hundreds of attoseconds depends on molecular structure. The method can be extended to other molecules where the HHS technique can be employed.
Ultrafast electron dynamics are of crucial importance for understanding and steering complex chemical and biological reactions. When subjected to an intense laser field, a pure electron-driven dynamic can be initiated by strong field ionization of the molecule. Theoretically, this dynamic manifests itself as a migration of the initially created hole density through the molecular backbone and was dubbed charge migration (CM) in 1999.1 In the last decade, CM has been extensively studied theoretically using time-dependent quantum chemistry packages.2–13 Topics of interest include the dependence of CM on molecular species and structures,9–13 as well as the role of nuclear dynamics and decoherence in CM.14–16 In these numerical studies, calculations were made to follow the time evolution of the density of the wave packet under the field-free condition. However, these results cannot be tested against experiments directly. In a real experiment, to know the field-free wave packet (or CM), a probe pulse is needed, which will inevitably affect the evolution of the wave packet. Thus, retrieving the field-free CM from the measurement is a monumental task and rarely possible.
Since it is a fascinating goal of ultrafast science to follow electron and/or nuclear dynamics, in spite of the difficulty, it is still desirable to perform pump–probe experiments to extract some partial information. For CM in molecules, it occurs at a timescale of a few tens of attoseconds. Then one would like to have pump and probe pulses shorter than a few femtoseconds. Such pulses, however, would have a bandwidth of a few tens of electron volts, and the created wave packet would be very complicated for studying valence electrons in a molecule. Additionally, the currently available attosecond pulses are still not intense enough for such applications. To date, most electron dynamics studies have used attosecond pump and intense IR probe pulses, or vice versa.17–21 Among the experiments that studied electron dynamics or CM, most of them are unable to obtain electron wave packets or the dynamics of CM for individual molecules directly.
From strong-field physics, it has long been suggested that one can use high-harmonic spectroscopy (HHS) to study attosecond electron dynamics using intense infrared lasers (see the reviews22,23). The principle of HHS is based on the three-step process of high-order harmonic generation (HHG),24,25 where an electron is first ionized near the peak of the laser field, the released electron then drifts in the laser field, and some portion will be driven back to recombine with the ion and emit harmonic radiation. At the time of recombination, the information of the electron wave packet of the cation is encoded in both the amplitude and phase of each generated harmonic. The intrinsic time-to-frequency mapping underlying HHG allows a temporal measurement of the electron dynamics with a resolution of tens to a hundred attoseconds.26–32 Up to now, HHS has been well used for ultrafast molecular detection including static molecular orbital tomography33–35 and also dynamic imaging of nuclear and electron motions.26–32
Sign up for Advanced Photonics TOC. Get the latest issue of Advanced Photonics delivered right to you!Sign up now
Although the HHS principle is easy to understand, implementing it for extracting accurate CM for each single molecule is not so straightforward, since the experimentally measured harmonic spectra are due to the coherent summation of the individual radiation weighted by the angular distribution of the molecules. In the first experiment of using HHS to extract the CM by Kraus et al.,26 the measured angularly integrated harmonic spectra were directly taken as the single-molecule results. In doing so, large errors are introduced into the reconstructed dynamics. In our recent paper,36 we developed a machine-learning (ML) algorithm to retrieve the single-molecule harmonics from the angularly integrated harmonic spectra, opening an avenue for accurate measurement of CM on the single-molecule level.
In spite of that, the sensitive dependence of CM on molecular orbitals and orientations still makes the CM dynamics complex and difficult to trace. There are still some open questions about CM that remain unclear; for example, how fast does the charge migrate in molecules? Very recently, by creating a localized wave packet, the hole has been predicted to migrate along the molecular backbone in a particle-like manner at a speed of a few Å/fs.10 However, in most reactions involving valence electrons, delocalized wave packets are created. In addition, the electron wave packet cannot be measured directly. They have to be reconstructed from other experimental data. In this work, we focus on a linear carbon chain molecule, butadiyne (), where the movement of the hole is expected to be along the carbon backbone.9 Using an ML algorithm to analyze the harmonic spectra measured from aligned in a two-color driving field, we successfully retrieved the hole wave packet of the cation in . From the center of charge (COC) at each time, the CM speed was measured for the first time. Our approach allows us to extract how migration speed depends on the alignment angle of the molecule, whether the charge density is localized10–12 or delocalized.
2 Results and Discussion
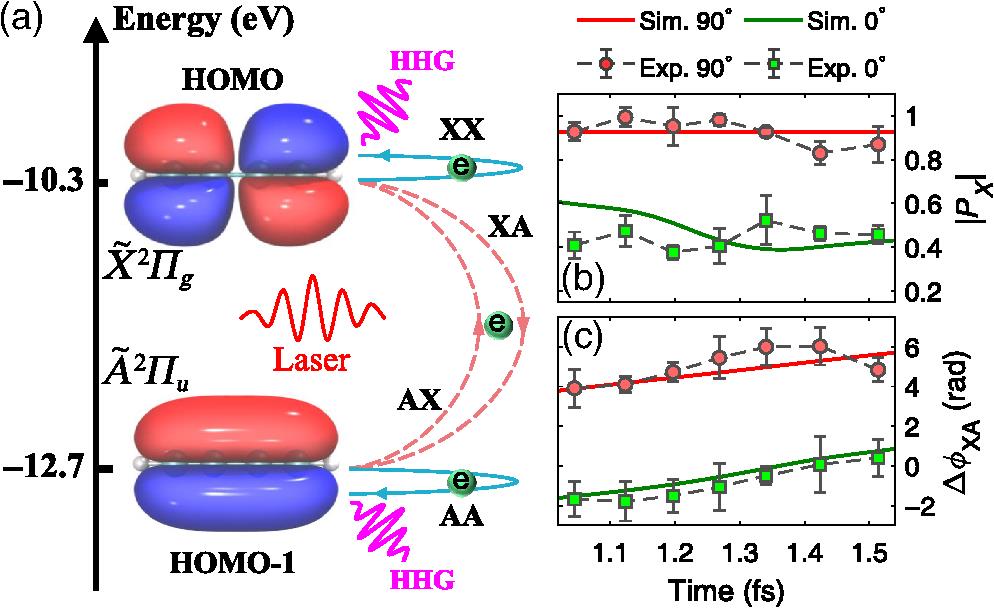
Figure 1 sketches the multichannel mechanism of HHG from the molecule. Exposed to an intense external laser field, the ground () and the first excited () states of the ion, which correspond to the removal of an electron from the highest and second-highest occupied molecular orbitals HOMO and HOMO-1, respectively, can be simultaneously populated by the strong-field ionization due to their close vertical ionization potentials (). The coherent superposition of multiple electronic states of the molecular ion creates a many-electron wave packet, which evolves in time, leading to the time-dependent variation of the charge density, i.e., the CM in the molecular ion. In the HHG process, these ionic states act as the intermediate that connects the same initial and final state of the system. The presence of different ionic states between the ionization and recombination provides different channels for the harmonic radiation.37–39 For , it includes two diagonal channels XX, AA, and two off-diagonal channels XA and AX, as shown in Fig. 1(a). The off-diagonal channels reflect the laser-induced coupling between the two ionic states during the HHG process. Eventual harmonic radiation is a coherent superposition of each channel,37–39 i.e., where is the total dipole moment for HHG, and is the dipole related to each channel, which in our reconstruction is calculated by the quantitative rescattering theory.40–43 is a complex mixing coefficient related to the laser-induced transitions between the two ionic states during the harmonic generation process. Equation (1) suggests that the electron dynamics is naturally recorded in the harmonic spectrum and can be deciphered by disentangling the multichannel contributions from the total dipole moment.
Figure 1.Probing CM in with HHS. (a) Schematic layout of the multichannel HHG in molecule that involves the ground () and first excited states () of the molecular ion. In , there are four channels labeled as XX, AA, XA, and AX, respectively, contributing to HHG. Here the first and second letters label the ionic state after ionization and before recombination, respectively. (b), (c) Experimentally retrieved population amplitude () of the state (b) and the relative phase () between the wave functions of and states (c) for the parallel (0 deg, green squares) and perpendicular (90 deg, red circles) alignment of the molecule. The solid lines show the TDDFT results for comparison. Error bars in panels (b) and (c) represent the SDs of the reconstructions, which are estimated from the experimental errors of the HHG signals with the bootstrap method.
In this work, we have carried out HHG experiment of using a commercial Ti:sapphire laser system (Astrella-USP-1K, Coherent, Inc.), which delivers 35 fs, 800 nm laser pulses at a repetition rate of 1 kHz. The output laser is split into two beams. One, with moderate intensity (aligning pulse) is used to create nonadiabatic alignment of molecule. The other intense one (probe pulse) is used to generate high-order harmonics. The aligning and probe pulses are parallel in the polarization (for more experimental details, see Supplementary Material). In our experiment, we first measured the HHG from molecule with a one-color driving scheme. The measured HHG signals at different time delays between the probe and aligning pulses were used to determine the alignment distribution in our experiment with the method reported in Ref. 44. With molecular alignment distribution determined, the ML algorithm developed in Ref. 36 then was used to analyze the delay-dependent HHG data to disentangle the coherent angular average of high harmonics originating from the imperfect molecular alignment in experiment, since this angular average could compromise the measurement from the real single-molecule response.45–47 This is especially important for a low degree of molecular alignment in our experiment, which has (see Fig. S2 in the Supplementary Material).
Second, to extract the CM dynamics, multiple experimental observables are required to decompose the multichannel contributions, since in the one-color laser field, only one set of can be obtained for each harmonic order, which is insufficient for the decomposition. Here we introduce a two-color driving scheme, where the laser field is synthesized by an intense 800 nm fundamental field and a weak second-harmonic (SH) field that has the same polarization direction as the fundamental one (for experimental details, see Supplementary Material) to generate high-order harmonics. In our experiment, the SH field used is weak ( of the fundamental field), which barely alters the electron dynamics of the molecular ion (see Supplementary Material), but could affect the harmonic radiation of each channel. The measurements at different relative phases of the two-color fields thus can replenish the data set required for the reconstruction. By applying the ML-based reconstruction procedure36 to the measurements at different two-color relative phases, we are able to obtain the complex mixing coefficients of the multiple orbitals of the molecular cation, for each fixed-in-space angle, at the instant of recombination when each harmonic order occurs. To ensure the one-to-one mapping between the instant time and harmonic order, the short electron trajectory has been selected by phase matching in the experiment.
Figure 1(b) shows the time-dependent population amplitudes of the ground state () of ion extracted from the experimental data of H11 to H17 (including both even and odd orders) for the molecules aligned at 0 deg (squares) and at 90 deg (circles) to the polarization of the driving field, respectively. Figure 1(c) displays the corresponding relative phases between the population coefficients of the and states (). In our reconstruction, the uncertainties of the retrieved parameters [error bars in Figs. 1(b) and 1(c)] were estimated from the experimental errors of the HHG signals with the bootstrap method.48 For comparison, we have simulated the above parameters based on the time-dependent density functional theory (TDDFT). In the TDDFT framework, the molecular system is described by a series of one-particle Kohn–Sham (KS) orbitals, in which the evolution can be obtained by solving the time-dependent Kohn–Sham (TDKS) equations. 49,50 The inclusion of laser-induced coupling between different orbitals in the TDKS equations allows us to simulate the evolution of different orbitals during the driving laser field. In our calculations, the TDKS equations are solved using the Octopus package51 with an local-density approximation (LDA) exchange-correlation functional52 and an average-density self-interaction correction.53 In our calculations, the molecule is assumed to align along the direction. The driving laser field is polarized in the plane and propagates along the direction. The TDKS equations are solved on a Cartesian grid with the size of a.u. The time step in the simulations is fixed at 0.05 a.u., and the spatial spacing is 0.38 a.u. To eliminate artificial reflections from boundaries, the wave function has been multiplied by a -masking function at each time step. With the time-dependent TDKS orbitals obtained, the transition amplitude can be calculated by , where is the initial KS orbital and the subscript denotes the and states involved in the CM dynamics. As shown in Figs. 1(b) and 1(c), the TDDFT results (solid lines) agree well with the experimental reconstructions.
With the retrieved parameters in Figs. 1(b) and 1(c), we can reconstruct the hole wave packet of the ion. We first examine the results for the 90 deg alignment of the molecules. In this case, by symmetry, the two states are not coupled by the laser, such that populations of the and states do not change with time; see the red line in Fig. 1(b). In this situation, the reconstructed charge density does change with time, as can be seen in Fig. 2(a), which displays the reconstructed hole densities in ion at the recombination times of H12 to H17. These figures also show substantial hole migration along the molecular backbone ( axis). Moreover, the hole densities are always symmetric about the plane because of the symmetries of the HOMO and HOMO-1 orbitals of . Note that these results are similar to the field-free CM defined by Cederbaum and Zobeley.1
Figure 2.Reconstruction of CM in for perpendicular alignment. (a) Snapshots of the reconstructed hole densities for the alignment angle of 90 deg. (b) Time-dependent hole densities along the molecular backbone obtained by integration over the direction. For clarity, the molecular backbone has been plotted on the top of panel (b). (c) Time-dependent COC position (dashed line with circles) retrieved from the hole densities in (b). Here the dashed-dotted line is a linear fitting of to evaluate the CM speed, and the green squares represent the SD of the coordinate. (d) Flux of charge density crossing the plane. Negative value means CM from side to side.
To provide simpler information from the charge density distribution, we extracted the reduced hole density by integrating the hole density over the direction. As shown in Fig. 2(b), the hole created by ionization is initially localized at the left triple bond, then spreads over the molecule, and finally is distributed around the right triple bond after about 500 as. To quantify the CM dynamics, we further define the expectation value of the abscissa (carbon chain) as the COC position. As shown in Fig. 2(c), the extracted COC position (blue circles) increases almost linearly with time, indicating a unidirectional migration of the hole from the side to the side. By a linear fitting of [the dashed-dotted line in Fig. 2(c)], we can then evaluate the CM speed from the slope, which is about Å. This result is close to the theoretical predictions in halogenated hydrocarbon chains.10 We have also calculated the standard deviation (SD) of the abscissa to quantify the degree of localization of the hole during the evolution. As shown in Fig. 2(c), the SD of (green squares) increases in the first 200 as and turns to decrease in the last 300 as, which reflects a delocalization-to-localization behavior of the created hole wave packet. The decrease is due to the wave packet beginning to bounce back from the right bond. At a longer time, the hole was predicted to travel back and forth between the triple bonds on both sides.9,10 Such a periodic behavior cannot be fully visualized from the reconstructions in Figs. 2(b) and 2(c) due to the limited temporal window in our measurement. With the retrieved CM speed, we can estimate the CM-mode period or frequency that is critical for describing the periodic dynamics, by , where a.u. is the distance between the centers of the two triple bonds in , which is approximately the CM distance in . For the quasi-field-free CM, the CM-mode period and frequency are estimated to be 1.76 fs and 2.35 eV, which are close to the results 1.72 fs and 2.4 eV defined by the energy difference between the two ionic states of .
Rigorously, single-hole dynamics are to be obtained with quantum mechanics. From the retrieved complex wave packet, real values of probability density and current density can be obtained, and they have been found to satisfy the equation of continuity, . In Fig. 2(d), we show the time-dependent total flux crossing the plane calculated from the retrieved complex hole wave packet. One can see that the total flux is always negative over the 500 as duration, indicating continuous migration of the charge from side to the side. A near-zero flux near 1.5 fs also reflects that the hole has moved to the right terminal bond. These results are consistent with the analysis in Fig. 2(c). On the other hand, the flux changes rapidly with time, indicating that the constant speed in Fig. 2(c) is valid only for the speed of the COC. From the other viewpoint, this would imply that itself, or the hole density alone, would not provide adequate information on electron dynamics except from the retrieved wave packet. Like electrons and holes in solids, could provide a first-order interpretation of carrier dynamics, even though the carriers are not localized. This is how the speed of CM is to be understood. It is a measure of how the average position moves.
Proceeding further, next, we consider in Fig. 3 the reconstructed results for the parallel alignment of the molecule. In this case, the ground and first excited states of are strongly coupled by the laser field due to the large transition dipole moment between these two states along this direction. The resulting CM is controlled by the laser field. Comparing Fig. 2 with Fig. 3, we can see that laser coupling would drive the hole density to side faster [Fig. 3(a)], as well as for the reduced hole density [Fig. 3(b)]. The COC position also rises faster, from which the CM speed has increased to Å, even though the SD remains roughly the same, except the feature changes at earlier times [Fig. 3(c)]. Note that here the CM speed is obtained by fitting the COC position before 1.34 fs, where increases almost linearly. Afterward, the hole reaches and localizes at the right terminal bond. In Fig. 3(d), the flux also shows faster changes with time, indicating that laser coupling has substantially changed the hole density distribution.
Figure 3.Reconstruction of CM in for parallel alignment. (a)–(d) The same as Fig. 2(a)–2(d), but for the case of parallel alignment of the molecule.
To evaluate the reconstructed hole dynamics, we have further performed TDDFT simulations. Figures 4(a) and 4(b) show the time-dependent hole density and the COC position simulated in the same temporal range as in the experiment (i.e., 1.04 to 1.51 fs) for 90 deg alignment of the molecule. One can see that the TDDFT results in Figs. 4(a) and 4(b) agree well with the experimental reconstructions presented in Figs. 2(b) and 2(c). The laser-controlled CM in the parallel aligned molecules has also been simulated. As shown in Figs. 4(c) and 4(d), under the influence of the laser field, the simulated CM dynamics are far different from the field-free result in Figs. 4(a) and 4(b). The simulated increases almost linearly before 1.34 fs and turns to decrease afterward. These results are also in reasonable agreement with the experimental reconstructions in Figs. 3(b) and 3(c).
Figure 4.TDDFT simulations of the CM dynamics in . (a), (b) TDDFT calculations of the reduced hole density and COC position in the experimental temporal range for the 90 deg alignment of the molecule. (c), (d) Same as panels (a) and (b), but for the parallel alignment (the alignment angle of 0 deg) of the molecule.
Using our retrieval method, we are able to obtain CM for all alignment angles at the same time. The CMs for other alignment angles of the fixed-in-space molecules have also been reconstructed (see Figs. S6 and S7 in the Supplementary Material). The CM speed retrieved as a function of the alignment angles is shown in Fig. 5 (squares). For comparison, the TDDFT results are also presented (solid line). The CM is demonstrated to depend sensitively on the alignment angles. Moreover, the speed of the laser-controlled CM (the cases of non-perpendicular alignments) is faster than the field-free result at 90 deg. The increase of the CM speed mainly arises from the laser-induced coupling between these two cation states. For the 90 deg alignment (field-free case), the timescale of the charge oscillations is determined by the intrinsic energy gap between the two cationic states. While for the 0 deg alignment (and other angles), laser-induced coupling between these two cation states gives rise to a much faster increase in the relative phase between these two states (see Fig. S9 in the Supplementary Material), which is equivalent to a larger energy gap between the two cationic states, thus yielding a much faster charge oscillation. This is similar to the well-known fact that the field-free two-level oscillation is slower than the Rabi oscillation when the two levels are driven by an intense laser pulse. In addition, it has been reported that the deviation of the experimentally extracted optimal two-color relative phases , where the HHG intensity reaches a maximum with respect to the single-active electron response will be a key indicator for the multichannel (CM) dynamics in the molecules.54 We have examined our two-color measurements and compared them to the single XX channel calculations. The results obtained at the alignment and anti-alignment of molecule are shown in Fig. S10 in the Supplementary Material. One can see a clear difference in the deviations of in the two cases, which provides direct experimental evidence of the different CM dynamics along these two orientations. The alignment dependence of the CM dynamics proves that one should not obtain the corresponding single-molecule dynamics directly from the experiment without angular deconvolution. On the other hand, it also provides a potential way to control the CM speed and even ultimately to manipulate the rate of a chemical reaction.
Figure 5.Reconstruction of the CM speed in . Dashed line with squares shows the CM speed retrieved as a function of the alignment angles. The solid line plots the TDDFT result for comparison.
Finally, it is worth mentioning that in our study of , there are only two cationic states involved in the CM dynamics. For other molecules, such as molecules,27,28 more cationic states will participate in the electronic wave packet. In such cases, one can anticipate more complex migration dynamics in the molecule. Due to the different symmetries of the cationic states, the resulting movement of the hole may no longer be solely along the molecular backbone. Instead, the hole could traverse across the molecular backbone or exhibit swirling motion around the constituent atoms.27,28 In such scenarios, a vertical component of the CM speed (along the direction) should be defined to comprehensively describe the complex migration dynamics in space.
3 Conclusions
In summary, CM in a carbon-chain molecule was measured using an ML-based two-color HHS method. The CM dynamics are reconstructed at the most fundamental level for each single fixed-in-space molecule. By analyzing the time-dependent hole densities, the speed of CM was extracted for the first time for each alignment angle of the molecule. The retrieval results demonstrate that the laser-controlled CM is much faster than the field-free one. These results are consistent with the TDDFT simulations. Our result provides a simple and intuitive way to understand and quantify the complex CM in molecules. Looking ahead, the method presented here may be extended to driving lasers with much longer wavelengths, in which the whole periodic hole migration may be directly observed from the measurement. On the other hand, oriented halogen-functionalized carbon-chain molecules are also ideal candidates for CM studies in experiments, where the influence of the hole localization on the CM speed may be examined. Moreover, the halogen functionalization can provide another degree of freedom to control the CM.
Lixin He is an associate professor at Wuhan National Laboratory for Optoelectronics, Huazhong University of Science and Technology (HUST), Wuhan, China. He received his PhD from HUST in 2016. From 2016 to 2018, he worked as a postdoctoral fellow in Prof. Peixiang Lu’s group at HUST. His main research interest is high-order harmonic generation and ultrafast imaging of molecular dynamics.
Yanqing He received his PhD in ultrafast optics, supervised by Prof. Pengfei Lan, from HUST in 2023. His research focuses on laser-induced molecular alignment and ultrafast molecular imaging.
Siqi Sun is a PhD student, supervised by Prof. Pengfei Lan, at HUST. His main research interest is AI algorithms and their applications in ultrafast molecular imaging.
Esteban Goetz is a postdoctoral research associate in theoretical atomic physics at the University of Connecticut, USA. He obtained his PhD from Universität Kassel, Germany. He also worked at Kansas State University and Drake University. His research activities and interests include laser-driven ultrafast quantum dynamics and quantum control, with a focus on high-harmonic generation and photoelectron spectroscopy.
Anh-Thu Le is a professor at University of Connecticut, USA. He received his PhD from Belarusian State University, Belarus. He also worked at Kansas State University and Missouri University of Science and Technology. His research interests include intense laser-atom/molecule interaction and attosecond physics, with main activities focusing on strong-field ionization, high-harmonic generation spectroscopy, and laser-induced electron diffraction.
Xiaosong Zhu is a professor at the School of Physics, HUST, Wuhan, China. He received his PhD from HUST in 2013. From 2018 to 2020, he received the Alexander von Humboldt Research Fellowship and worked at Leibniz Universität Hannover, Germany. His research focuses on high harmonic generation from atoms, molecules, and solids.
Pengfei Lan is a professor at the School of Physics, HUST, Wuhan, China. He received his PhD from HUST in 2009. From 2009 to 2013, he worked at RIKEN, Japan. He was selected as a distinguished young scholar of NSFC. His research focuses on the generation of attosecond pulses and high harmonics with atoms, molecules, and solids.
Peixiang Lu is a professor and the vice director at Wuhan National Laboratory for Optoelectronics, HUST, Wuhan, China. He received his PhD from Shanghai Institute of Optics and Fine Mechanics. He was selected as a Cheung Kong Scholar Chair Professor and a distinguished young scholar of NSFC. He was selected as OSA fellow in 2016. His research interest is strong ultrafast optics.
Chii-Dong Lin received his PhD in physics in 1974 from the University of Chicago under Prof. Ugo Fano. He has been a professor at Kansas State University since 1976. His research spans many fields of AMO physics. Since 2002, he has contributed several ideas in strong-field physics, including the MO-ADK theory for ionization and the QRS model for electron rescattering and harmonic generation. He also contributed to the characterization of attosecond pulses and dynamics of molecules from intense laser experiments.