
Jiani Lin, Fangxu Wang, Qi Rui, Jianfu Li, Qinglin Wang, Xiaoli Wang. A novel square planar N42− ring with aromaticity in BeN4[J]. Matter and Radiation at Extremes, 2022, 7(3): 038401
- Matter and Radiation at Extremes
- Vol. 7, Issue 3, 038401 (2022)
Abstract
I. INTRODUCTION
Polymeric nitrogen containing single and double nitrogen bonds releases a large amount of energy during its decomposition and is thus a candidate green high-energy-density material (HEDM). However, the synthesis of stable HEDMs containing breakable single or double nitrogen bonds poses a real challenge. To date, there has been experimental confirmation of four polymeric nitrogen structures: cg-N (synthesized at 2000 K and 110 GPa),1 LP-N (3000 K and 150 GPa),2 HLP-N (3300 K and 244 GPa),3 and bp-N (2200 K and 146 GPa).4 However, these structures are synthesized under high temperatures and pressures, which limits their application as HEDMs. Therefore, it is an urgent, although formidable, challenge to explore novel polymeric nitrogen structures that can be synthesized under mild conditions.
Numerous theoretical and experimental results have indicated that the addition of metallic elements (M) to the nitrogen materials could provide new compound forms of polymeric nitrogen (MNx) at lower pressures. Compared with pure polymeric nitrogen structures, these may have higher stability and milder synthesis conditions. Charge transfer from metal to polynitrogen may play important roles in reducing the pressure required for synthesis and improving structural stability. Theoretical calculations have predicted a variety of polynitride compounds MNx (M = Li, Be, Na, Mg, Al, K, Ca, Cs, Rb, Ba, Se, He, or Xe, with x = 3, 5, 6, 10) featuring a variety of N5 or N6 polymeric nitrogen chains5–30 and some complicated nitrogen clusters [fused N18 rings in KN8 (Ref. 8) and N10 rings in BeN4 (Ref. 29), etc.]. The transition metals (Sc,31,32 Mo,33 Tc,34 Re,35 Fe,36,37 Ru,38 Os,39 Rh,40 Ir,39 Pd,41 Pt,41 Cu,42 Ag,43 Au,44 Zn,45 and Hg46) can also form nitrogen-containing structures. Some of these compounds are not potential HEDMs, but they do prove that introducing metallic elements to nitrogen seems to be a feasible way to enhance metastability. Experimentally, some metals or their compounds have been used as raw materials to synthesize polynitrogen compounds at relatively low pressures. CsN5 has been successfully synthesized by compressing CsN3 with N2 near 60 GPa,47 and solid LiN5 solid has been obtained by compression and laser heating of Li embedded in molecular N2 at 45 GPa.48 Fe3N2, FeN2, and FeN4 have been synthesized from Fe and N2 under different pressures,49 and elemental Cu reacts with N2 to form CuN2 at 50 GPa.50 Very recently, a beryllium tetranitride tr-BeN4 in the triclinic phase has been synthesized at 85 GPa,51 and it has similar structural units as the previously predicted P-1 BeN4 (infinite N chains coordinating Be atoms), proving that crystal structure prediction (CSP) searches can provide credible results.
The alkaline earth metals Be, Mg, Ca, Sr, and Ba are adjacent to the alkali metals in the Periodic Table, with similar properties but with a wider scope for chemical reactions and compound formation because of their additional valence electron. Among the alkaline earths, Be has the lowest atomic weight, and therefore its compounds with nitrogen have a high mass density of N and are good candidate HEDMs. A layered material, BeN4, containing corrugated N10 rings has recently been synthesized.51 Such a single-bonded polymeric nitrogen network has been predicted theoretically.29,52 Therefore, we have used an unbiased structural search with a particle-swarm optimization (PSO) algorithm53,54 to predict new beryllium nitrides with 1:4 compositions and have thereby found a new BeN4 compound that may be stable at ambient pressure. In this compound, a planar N42− ring is surrounded by four Be atoms, and the six π electrons of this ring conform to the Hückel [4n + 2] rule (with n = 1), indicating aromaticity of BeN4. The large mass ratio of nitrogen in this compound (86.15%) leads to a high energy density (3.924 kJ/g).
II. COMPUTATIONAL DETAILS
We used the Crystal Structure Analysis by Particle Swarm Optimization (CALYPSO) code53,54 to search the structures of BeN4 at different pressures, namely, 0, 10, 20, 50, and 100 GPa, at 0 K. Geometrical optimizations, total energy calculations, and electronic structure calculations were carried out in the framework of density functional theory (DFT) using the generalized gradient approximation with the Perdew–Burke–Ernzerhof functional (GGA-PBE),55 with a cutoff energy of 500 eV and a 0.03 Å−1k-point grid56 in the Vienna Ab initio Simulation Package (VASP).57 The convergence threshold was used to optimize the atomic positions and lattice constants until the forces on each atom were less than 0.01 eV/Å and the energy change was less than 1.0 × 10−6 eV/atom. Projector augmented wave (PAW)58 pseudopotentials were adopted for Be and N, with 1s22s2 and 2s22p3 valence states, respectively. Phonon dispersion relations were calculated using a supercell method with the finite displacement approach59 through the Phonopy code.60Ab initio molecular dynamics (AIMD) simulations were performed within the NVE ensemble61,62 to study thermal stability. The strain–stress methodology was used to calculate the elastic constants. The bulk and shear modulus, Young’s modulus E, and Poisson’s ratio v were obtained from the Voigt–Reuss–Hill approximations.63 The detonation velocity and pressure were calculated through the Kamlet–Jacobs semiempirical equations Vd = 1.01(NM0.5Ed0.5)0.5(1 + 1.30ρ) and Pd = 15.58ρ2NM0.5Ed,64,65 where N is the number of moles of gas per gram, M is the molar mass of the N2 gas (28 g/mol), Ed is the energy density, and ρ is the mass density of BeN4. The crystal orbital Hamilton population (COHP) was calculated to characterize the bonding properties using the LOBSTER program.66 Charge transfer was based on a Bader analysis,56,67 and the chemical bonding mode was calculated by the Solid State Adaptive Natural Density Partitioning (SSAdNDP) method.68
III. RESULTS AND DISCUSSION
A. Geometric structures
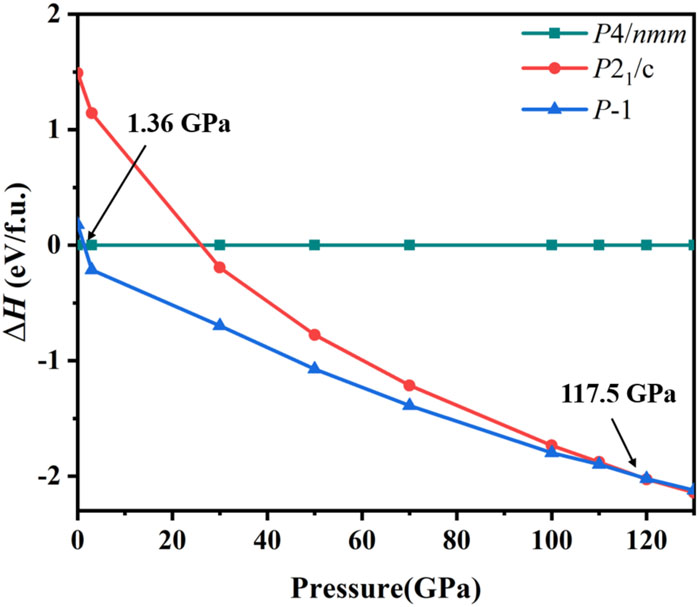
In a previous study, Zhang et al.29 used an unbiased structure search method to explore the structural evolutionary behaviors of beryllium polynitride compounds below 200 GPa. Interestingly, the only stable N-rich composition was found to be BeN4, with the compounds P1-BeN4 and P21/c-BeN4 being theoretically predicted to exist at high pressure. In our work, also using an unbiased structure search method with CALYPSO, we have predicted a new BeN4 in the tetragonal phase with the high group symmetry of P4/nmm. The calculated enthalpies of these three structures at pressures from 0 to 130 GPa at T = 0 K are shown in Fig. 1.
![]()
Figure 1.Enthalpy difference of BeN4 relative to the
BeN4 undergoes structural phase transitions first from P4/nmm to P1 at a pressure of 1.36 GPa and then to P21/c at 117.5 GPa. The phase transition pressure from P1 to P21/c phase that we calculated is similar to the reported value (115.1 GPa) in Fig. 1, which indicates that our method is appropriate. The predicted crystal structure of P4/nmm and its corresponding lattice parameters and atomic coordinates are shown in Fig. 2 and Table I, respectively. According to Fig. 2, there is a square planar N42− ring in P4/nmm-BeN4, where each inner N atom is shared by one Be atom and two adjacent N atoms. Within the cyclo-N4, the length of the N–N bond is 1.363 Å and the angles of the N–N–N bonds are all around 90° at ambient pressure. Be atoms are situated at the center or vertex positions, with each Be atom binding to four adjacent N atoms with equal bond lengths of 1.713 Å. A variety of N4 clusters have been predicted theoretically,24,30,69–71 including N44−, N42+, N42−, and N4+. However, the novel square planar N42− ring predicted in the present work has not previously been reported, although a square planar N42− ring was found in bipyramidal Li2N4 by van Zandwijk et al.,72 and a series of alkali metal compounds M2N4 and alkaline earth compounds MN4 with D4h bipyramidal, D2h planar, and C2v planar structures were also found to contain such an N42− ring.70,71,73 Here, we shall now focus on the square planar N42− ring in P4/nmm-BeN4.
![]()
Figure 2.Crystal structure of the stable
| Space group | Pressure (GPa) | Lattice parameter (Å°) | Atom | Wyckoff position | x | y | z | |
|---|---|---|---|---|---|---|---|---|
| P4/nmm | 0 | a = 5.03 | α = 90.00 | Be | 4e | 0.500 | 0.500 | 0.500 |
| b = 5.03 | β = 90.00 | N | 16k | 0.000 | 0.692 | 0.327 | ||
| c = 4.21 | γ = 90.00 | |||||||
Table 1. Lattice parameters and atomic positions for P4/nmm-BeN4 at 0 GPa.
B. Kinetic and thermodynamic stability
Because there are no imaginary frequencies in the entire Brillouin zone (Fig. 3), the calculated phonon dispersions show that P4/nmm-BeN4 is dynamically stable at ambient pressure. In addition, the phonon density of states shows that the low-frequency vibrational mode can be mostly attributed to the strong coupling vibration between Be and N atoms, while the N–N stretching mode is responsible for the high-frequency vibrational mode.
![]()
Figure 3.Phonon dispersions and phonon density of states (PHDOS) of
We further explore the thermal stability of BeN4 at ambient pressure and high temperature by performing AIMD simulations on a 3 × 2 × 3 supercell containing 180 atoms. The images of the geometrical structure in Figs. 4(a) and 4(b) clearly reveal that the square planar N42− ring maintains its structural integrity without any visible distortion after heating at 300 or 500 K for 10 ps with a time step of 1 fs. The radial distribution function (RDF) of N–N separations in P4/nmm-BeN4 is shown in Fig. 4(c). The first sharp RDF peak is the average distance between the selected atom and its nearest surrounding atoms, here corresponding to the N–N bonds of BeN4. This indicates that the N–N bond lengths remain at 1.369 Å as the temperature rises from zero to 500 K, which proves the stability of P4/nmm-BeN4 at high temperatures. Thus, BeN4 has a moderate cohesive energy and good thermal stability.
![]()
Figure 4.Molecular dynamics simulations and radial distribution function of
C. Mechanical stability
We also calculated the elastic constants of the P4/nmm-BeN4 structure because these are helpful for determining the mechanical stability and hardness of a material. The elastic constants Cij, bulk modulus B, shear modulus G, the Young’s modulus E and Poisson’s ratio v of P4/nmm-BeN4 are presented in Table II. They are related by E = 9BG/(3B + G) and v = (3B − 2G)/[2(3B + G)].74 We calculated the bulk modulus of the β-Be3N2 hexagonal structure at ambient pressure to be B = 237.18 GPa, which is similar to a previously published value for this compound (236 GPa),75 which indicates that the calculation method we have adopted is appropriate for the Be–N system. The Pugh ratio k = B/G is used to estimate ductile or brittle characteristics, with k < 1.75 corresponding to brittle behavior and k ≥ 1.75 to ductile behavior. The value of k for the P4/nmm-BeN4 structure is 1.02, indicating that this compound is brittle. For a tetragonal structure to be stable, the Cij should satisfy the Born–Huang stability criteria C11 > 0, C33 > 0, C44 > 0, C66 > 0, C11 − C12 > 0, C11 + C33 − 2C13 > 0, and 2(C11 + C12) + C33 + 4C13 > 0. The elastic constants of P4/nmm-BeN4 satisfy these criteria, indicating that it should be mechanically stable under ambient conditions.
| P4/nmm | Be3N2 | ||
|---|---|---|---|
| Elastic stiffness constants Cij | C11 | 224.82 | 517.77 |
| C22 | |||
| C33 | 20.82 | 479.98 | |
| C44 | 111.15 | 190.16 | |
| C55 | 2.29 | ||
| C66 | 2.29 | 185.58 | |
| C12 | 6.21 | 137.45 | |
| C13 | 20.32 | 87.57 | |
| B (GPa) | 41.75 | 237.18 | |
| G (GPa) | 41.06 | 192.14 | |
| E (GPa) | 92.78 | 453.87 | |
| v | 0.13 | 0.18 | |
| k | 1.02 |
Table 2. Elastic constants of P4/nmm-BeN4 and Be3N2 at ambient pressure.
D. Explosive performance
Energy density Ed is important for energy storage capability, while detonation velocity Vd and pressure Pd are known to be significant indices of detonation performance. The dissociation energy of P4/nmm-BeN4 is calculated for the following decomposition path under ambient conditions: 3BeN4(s) → Be3N2(s) + 5N2(g). We use Ed = (96.4853 × BeN4 molecule energy released)/(molar mass of BeN4) to calculate the energy density of P4/nmm-BeN4, the calculated Ed is about 3.92 kJ/g with reference to the lowest-energy phase at ambient pressure29,76 (the phase transition in Fig. S1 in the
| Compound | ρ (g/cm3) | Ed (kJ/g) | Vd (km/s) | Pd (kbar) |
|---|---|---|---|---|
| P4/nmm-BeN4 | 1.01 | 3.92 | 7.35 | 158.11 |
| TNT | 1.64 | 4.30 | 6.90 | 190.00 |
| HMX | 1.90 | 5.70 | 9.10 | 393.00 |
Table 3. Detonation properties of P4/nmm-BeN4, TNT, and HMX.
E. Electronic properties
To illustrate the electronic properties of BeN4, its band structure, projected density of states (PDOS), and crystal orbital Hamilton populations (COHPs) under ambient conditions are presented in Fig. 5. There are no bands crossing the Fermi level in the band structure in Fig. 5(a), which indicates that the predicted BeN4 crystal is a semiconductor with a bandgap of 3.67 eV. The obvious overlap between Be 2p and N 2p in the PDOS [Fig. 5(b)] reveals that the orbitals are strongly hybridized. Therefore, the bonding between Be and N is partially covalent. To characterize the orbital interaction between two N atoms, we performed a COHP analysis to measure the overlap strength between two N atoms. Negative values correspond to bonding states and positive values to antibonding states in Fig. 5(c), where it can be seen that the bonding states between N and N in BeN4 are completely occupied and the antibonding states partially occupied, resulting in strong covalent bonding. The integrated COHPs (ICOHPs) calculated from the wave function’s energy-weighted population between two atomic orbitals represent the bonding strength. The ICOHP values of N–N and Be–N bonds are −12.21 and −1.63 eV, respectively. Therefore, the N–N interaction is substantially stronger than the Be–N interaction, which indicates that the covalent bonding between Be and N is weaker, which is consistent with the results of our previous hybridization investigation. Moreover, the Bader charges of Be and N atoms shown in Table S1 in the
![]()
Figure 5.Electronic properties of
F. Chemical bonding pattern
We analyzed the chemical bonding patterns in the P4/nmm-BeN4 crystal using SSAdNDP software. In a primitive cell, Be atoms connect four neighboring N4 rings via four two-center, two-electron (2c-2e) Be–N σ bonds [Fig. 6(a)], and four N atoms form a ring through four 2c-2e N–N σ bonds [Fig. 6(b)]. There are two types of π orbital [Figs. 6(c) and 6(d)]: one links two adjacent N atoms through two 2c-2e N–N π bonds, while the other links four N atoms via one four-center, two-electron (4c-2e) N–N large π bond in a plane. Together, the σ and π bonds require 2 × 11 = 22 electrons, which is equal to the number of outer valence electrons of BeN4. It can be seen that the localized 2c-2e σ bonds, 2c-2e π bonds, and delocalized 4c-2e π bonds have an influence on the high structural stability of BeN4. The total of six π electrons is the most striking feature, since it satisfies the Hückel [4n + 2] rule (with n = 1), indicating that BeN4 is an aromatic structure.
![]()
Figure 6.SSAdNDP-derived chemical bonding pattern of
The band structure, PDOS, COHP calculations, and chemical bonding pattern analysis illustrate the covalent bonding between the N atoms. The Be atoms contribute their outer-shell electrons to the N4 cluster and use the outer-shell 2p orbital to accommodate the lone-pair electrons of N42−, with the formation of covalent and ionic bonds to stabilize the crystal. This explains why Be can stabilize the square N4 ring.
G. Synthetic route to square planar N42− ring
Be3N2 with two known phases (α80,81 and β82) has been experimentally synthesized. We have calculated the enthalpy of formation of the predicted P4/nmm-BeN4 relative to Be3N2 and N2 (the phase transition in Fig. S1 in the
![]()
Figure 7.Enthalpy of formation of
IV. CONCLUSIONS
Through structural searches and density functional calculations under ambient conditions, we have found a P4/nmm-BeN4 structure with high kinetic and thermodynamic stability. The phonon dispersion and electronic properties of this structure indicate the presence of covalent bonding between two N atoms, with covalent and ionic bonding between Be2+ and square planar N42− enhancing the stability of the semiconductor BeN4 structure. The total of six π electrons satisfies the Hückel [4n + 2] rule (with n = 1), indicating that BeN4 has an aromatic structure. Furthermore, the energy density of P4/nmm-BeN4 is 3.92 kJ/g, making it a candidate high-energy-density material. The calculated formation of enthalpy of BeN4 indicates that the square planar N42− ring might be synthesized through mixing Be3N2 and N2 at 31.6 GPa. The results of this investigation of the role of novel chemical bonds in nitrogen chemistry should encourage experimental efforts to synthesize these promising high-energy materials.
SUPPLEMENTARY MATERIAL
ACKNOWLEDGMENTS
Acknowledgment. This work was supported by the National Natural Science Foundation of China under Grant Nos. 11974154, 11674144, and 11604133, and the Natural Science Foundation of Shandong Province under Grant Nos. ZR2018MA038, 2019GGX103023, Z2018S008, and 2019KJJ019.
References
[1] R.Academy, R.Boehler, D. A.Dzivenko, M. I.Eremets, A. G.Gavriliuk, I. A.Trojan. Single-bonded cubic form of nitrogen. Nat. Mater., 3, 558-563(2004).
[2] M.Kim, J.Smith, D.Tomasino, C. S.Yoo. Pressure-induced symmetry-lowering transition in dense nitrogen to layered polymeric nitrogen (LP-N) with colossal Raman intensity. Phys. Rev. Lett., 113, 205502(2014).
[3] G.Geneste, D.Laniel, P.Loubeyre, M.Mezouar, G.Weck. Hexagonal layered polymeric nitrogen phase synthesized near 250 GPa. Phys. Rev. Lett., 122, 066001(2019).
[4] A. A.Adeleke, H.Gou, C.Ji, B.Li, W.Liu, H. K.Mao, W. L.Mao, Y.Meng, V. B.Prakapenka, G.Shen, J. S.Smith, B.Wan, L.Yang, Y.Yao. Nitrogen in black phosphorus structure. Sci. Adv., 6, eaba9206(2020).
[5] X.Peng, B.Wei, Q.Wei, H.Yan, M.Zhang, C.Zhao. New stable structures of HeN3 predicted using first-principles calculations. J. Alloys Compd., 800, 505-511(2019).
[6] K.Bao, T.Cui, D.Duan, D.Li, B.Liu, Z.Liu, F.Tian, W.Wang, S.Wei, H.Yu. Bonding properties of aluminum nitride at high pressure. Inorg. Chem., 56, 7494-7500(2017).
[7] T.Cui, D.Duan, D.Li, Y.Liu, Z.Liu, F.Tian, S.Wei. Nitrogen-rich GaN5 and GaN6 as high energy density materials with modest synthesis condition. Phys. Lett. A, 383, 125859(2019).
[8] I. I.Oleynik, B. A.Steele. Novel potassium polynitrides at high pressures. J. Phys. Chem. A, 121, 8955-8961(2017).
[9] H.Liu, Q.Wei, H.Yan, M.Zhang. A new high-pressure polymeric nitrogen phase in potassium azide. RSC Adv., 5, 11825-11830(2015).
[10] H.Dong, A. R.Oganov, G.Qian, Y.Shen, J.Zhang, Z.Zhou, Q.Zhu. Novel lithium-nitrogen compounds at ambient and high pressures. Sci. Rep., 5, 14204(2015).
[11] Y.Cai, P.Hou, D.Li, L.Lian, B.Liu, B.Wang, S.Wei. Structural phase transition and bonding properties of high-pressure polymeric CaN3. RSC Adv., 8, 4314-4320(2018).
[12] I. I.Oleynik, B. A.Steele, A. S.Williams. Novel rubidium poly-nitrogen materials at high pressure. J. Chem. Phys., 147, 234701(2017).
[13] T.Cui, D.Duan, D.Li, X.Li, B.Liu, Z.Liu, F.Tian, S.Wei. Alkaline-earth metal (Mg) polynitrides at high pressure as possible high-energy materials. Phys. Chem. Chem. Phys., 19, 9246-9252(2017).
[14] J.Lv, Y.Ma, Y.Wang, L.Zhang. Materials discovery at high pressures. Nat. Rev. Mater., 2, 17005(2017).
[15] T.Cui, D.Duan, D.Li, Y.Liu, F.Tian, H.Wang, W.Wang, H.Yu. High-pressure bonding mechanism of selenium nitrides. Inorg. Chem., 58, 2397-2402(2019).
[16] Y.Ma, F.Peng, H.Wang, Y.Wang, Y.Zhang. Stable xenon nitride at high pressures. Phys. Rev. B, 92, 094104(2015).
[17] G.Frapper, B.Huang. Barium–nitrogen phases under pressure: Emergence of structural diversity and nitrogen-rich compounds. Chem. Mater., 30, 7623-7636(2018).
[18] S.Guo, Q.Jiang, J.Li, J.Lin, X.Wang, H.Zhu, Z.Zhu. Stable zigzag and tripodal all-nitrogen anion N44− in BeN2. AIP Adv., 9, 055116(2019).
[19] H.Liu, Y.Ma, F.Peng, Y.Yao. Crystalline LiN5 predicted from first-principles as a possible high-energy material. J. Phys. Chem. Lett., 6, 2363-2366(2015).
[20] P. J.Brothers, W.Henderson, L. C.Perera, P. G.Plieger, O.Raymond. Advances in beryllium coordination chemistry. Coord. Chem. Rev., 352, 264-290(2017).
[21] R.Frankovsky, G. M.Friederichs, M.Mangstl, J.Schmedt auf der Günne, S. B.Schneider, W.Schnick. Electronic and ionic conductivity in alkaline earth diazenides MAEN2 (MAE = Ca, Sr, Ba) and in Li2N2. Chem. Mater., 25, 4149-4155(2013).
[22] R.Frankovsky, S. B.Schneider, W.Schnick. Synthesis of alkaline earth diazenides MAEN2 (MAE = Ca, Sr, Ba) by controlled thermal decomposition of azides under high pressure. Inorg. Chem., 51, 2366-2373(2012).
[23] G.Frapper, B.Huang, A. R.Oganov, S.Yu, Q.Zeng, L.Zhang. Emergence of novel polynitrogen molecule-like species, covalent chains, and layers in magnesium-nitrogen Mg
[24] Y.Han, H.Liu, F.Peng, Y.Yao. Exotic stable cesium polynitrides at high pressure. Sci. Rep., 5, 16902(2015).
[25] L.Chen, Z.Hu, J.Li, X.Wang, N.Xu, H.Zhu. Layered polymeric nitrogen in RbN3 at high pressures. Sci. Rep., 5, 16677(2015).
[26] L.Chen, J.Li, H.Lin, X.Wang, H.Zhu. Polymerization of nitrogen in cesium azide under modest pressure. J. Chem. Phys., 141, 044717(2014).
[27] J.Botana, L.Chen, T.Cui, J.Li, H.Liu, M.Miao, X.Wang, M.Zhang, H.Zhu. Polymerization of nitrogen in lithium azide. J. Chem. Phys., 139, 164710(2013).
[28] X.Feng, J.Hao, W.Lei, Y.Li, D.Liu, H.Liu, Y.Ma, S. A. T.Redfern. Route to high-energy density polymeric nitrogen
[29] L.Liu, G.Yang, S.Zhang, Z.Zhao. Pressure-induced stable BeN4 as a high-energy density material. J. Power Sources, 365, 155-161(2017).
[30] T.Gao, H.Liu, A.Majumdar, F.Peng, Y.Yao, S.Zhu. Stable calcium nitrides at ambient and high pressures. Inorg. Chem., 55, 7550-7555(2016).
[31] J.Li, J.Lin, D.Peng, Q.Wang, X.Wang, H.Zhu. Stable nitrogen-rich scandium nitrides and their bonding features under ambient conditions. Phys. Chem. Chem. Phys., 23, 6863-6870(2021).
[32] M. A.Aslam, Z. J.Ding. Prediction of thermodynamically stable compounds of the Sc–N system under high pressure. ACS Omega, 3, 11477-11485(2018).
[33] L.-P.Ding, X.-F.Huang, C.Lu, P.Shao, F.-H.Zhang. Prediction of molybdenum nitride from first-principle calculations: Crystal structures, electronic properties, and hardness. J. Phys. Chem. C, 122, 21039-21046(2018).
[34] T.-T.Bai, H.-Y.Yan, Y.-Q.Yuan, G.-T.Zhang, Y.-R.Zhao, B.-B.Zheng. First-principles investigations of the structure and physical properties for new TcN crystal structure. Mol. Phys., 114, 1952-1959(2016).
[35] B.Wei, Q.Wei, H.Yan, M.Zhang, C.Zhao. High-pressure phases and pressure-induced phase transition of MoN6 and ReN6. Phys. Lett. A, 383, 2429-2435(2019).
[36] X.Cai, Y.Chen, H.Wang, H.Wang, H.Wang. Novel triadius-like N4 specie of iron nitride compounds under high pressure. Sci. Rep., 8, 10670(2018).
[37] P.Chen, F.Gao, N.Gong, H.Gou, H.Liu, T.Shen, R.Tian, B.Wan, L.Wu, Y.Yao. Prediction of stable iron nitrides at ambient and high pressures with progressive formation of new polynitrogen species. Chem. Mater., 30, 8476-8485(2018).
[38] M.Hasegawa, T.Kikegawa, S.Muto, K.Niwa, K.Soda, K.Suzuki, K.Tatsumi. Discovery of the last remaining binary platinum-group pernitride RuN2. Chem. - Eur. J., 20, 13885-13888(2014).
[39] E.Gregoryanz, R. J.Hemley, H. K.Mao, C.Sanloup, S.Scandolo, A. F.Young. Synthesis of novel transition metal nitrides IrN2 and OsN2. Phys. Rev. Lett., 96, 155501(2006).
[40] D.Dzivenko, M.Eremets, M.Hasegawa, K.Niwa, R.Riedel, K.Suzuki, I.Troyan. High pressure synthesis of marcasite-type rhodium pernitride. Inorg. Chem., 53, 697-699(2014).
[41] J. C.Crowhurst, C. L.Evans, J. L.Ferreira, A. F.Goncharov, P. G.Morrall, A. J.Nelson, B.Sadigh. Synthesis and characterization of the nitrides of platinum and iridium. Science, 311, 1275-1278(2006).
[42] J.Li, M.Miao, L.Sun, X.Wang, H.Zhu. Simple route to metal
[43] R.Dinnebier, M.Jansen, C. L.Schmidt, U.Wedig. Crystal structure and chemical bonding of the high-temperature phase of AgN3. Inorg. Chem., 46, 907-916(2007).
[44] P. R.Briddon, M. R. C.Hunt, S.Krishnamurthy, M.Montalti, M. J.Shaw, L.?iller, K.Svensson, M. G.Wardle. Nitrogen ion irradiation of Au(110): Photoemission spectroscopy and possible crystal structures of gold nitride. Phys. Rev. B, 70, 045414(2004).
[45] G.Jiang, T.Song, X. W.Sun, J. H.Tian, T.Wang. Theoretical investigation on the high-pressure physical properties of ZnN in cubic zinc blende, rock salt, and cesium chloride structures. J. Phys. Chem. Solids, 110, 70-75(2017).
[46] S.Guo, J.Li, J.Lin, Q.Wang, X.Wang, H.Wu, H.Zhu. High-pressure stable phases in mercury azide. Comput. Mater. Sci., 169, 109147(2019).
[47] J. C.Crowhurst, I. I.Oleynik, V. B.Prakapenka, E.Stavrou, B. A.Steele, J. M.Zaug. High-pressure synthesis of a pentazolate salt. Chem. Mater., 29, 735-741(2017).
[48] G.Gaiffe, G.Garbarino, D.Laniel, P.Loubeyre, G.Weck. High-pressure synthesized lithium pentazolate compound metastable under ambient conditions. J. Phys. Chem. Lett., 9, 1600-1604(2018).
[49] I. A.Abrikosov, G.Aprilis, M.Bykov, E.Bykova, I.Chuvashova, N.Dubrovinskaia, L.Dubrovinsky, K.Glazyrin, E.Koemets, I.Kupenko, H. P.Liermann, C.McCammon, M.Mezouar, A. V.Ponomareva, V.Prakapenka, F.Tasnádi. Fe-N system at high pressure reveals a compound featuring polymeric nitrogen chains. Nat. Commun., 9, 2756(2018).
[50] J.Binns, P.Dalladay-Simpson, M.-E.Donnelly, E.Gregoryanz, A.Hermann, R. T.Howie, M.Pe?a-Alvarez, M.Wang. Direct reaction between copper and nitrogen at high pressures and temperatures. J. Phys. Chem. Lett., 10, 1109-1114(2019).
[51] A. I.Abrikosov, I. A.Abrikosov, T.Bin Masood, M.Bykov, S.Chariton, N.Dubrovinskaia, L.Dubrovinsky, T.Fedotenko, K.Glazyrin, A. F.Goncharov, M.Hanfland, I.Hotz, M. I.Katsnelson, D.Laniel, M. F.Mahmood, A. V.Ponomareva, V. B.Prakapenka, A. N.Rudenko, J. S.Smith, F.Tasnádi. High-pressure synthesis of Dirac materials: Layered van der Waals bonded BeN4 polymorph. Phys. Rev. Lett., 126, 175501(2021).
[52] K.Bao, T.Cui, D.Duan, D.Li, B.Liu, Z.Liu, F.Tian, W.Wang, S.Wei. A novel polymerization of nitrogen in beryllium tetranitride at high pressure. J. Phys. Chem. C, 121, 9766-9772(2017).
[53] J.Lv, Y.Ma, Y.Wang, L.Zhu. CALYPSO: A method for crystal structure prediction. Comput. Phys. Commun., 183, 2063-2070(2012).
[54] J.Lv, Y.Ma, Y.Wang, L.Zhu. Crystal structure prediction via particle-swarm optimization. Phys. Rev. B, 82, 094116(2010).
[55] K.Burke, M.Ernzerhof, J. P.Perdew. Generalized gradient approximation made simple. Phys. Rev. Lett., 77, 3865-3868(1996).
[56] H. J.Monkhorst, J. D.Pack. ‘Special points for Brillouin-zone integrations’—A reply. Phys. Rev. B, 16, 1748-1749(1977).
[57] J.Furthmüller, G.Kresse. Efficient iterative schemes for
[58] P. E.Bl?chl. Projector augmented-wave method. Phys. Rev. B, 50, 17953-17979(1994).
[59] Y.Kawazoe, Z. Q.Li, K.Parlinski. First-principles determination of the soft mode in cubic ZrO2. Phys. Rev. Lett., 78, 4063-4066(1997).
[60] F.Oba, I.Tanaka, A.Togo. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B, 78, 134106(2008).
[61] S.Nosé. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys., 81, 511-519(1984).
[62] W. G.Hoover. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A, 31, 1695-1697(1985).
[63] R.Hill. Related content the elastic behaviour of a crystalline aggregate. Proc. Phys. Soc., London, Sect. A, 65, 349-354(1952).
[64] Y.Chang, Q.Zhang. Prediction of detonation pressure and velocity of explosives with micrometer aluminum powders. Cent. Eur. J. Energ. Mater., 9, 77-86(2012).
[65] J. E.Ablard, M. J.Kamlet. Chemistry of detonations. II. Buffered equilibria. J. Chem. Phys., 48, 36-42(1968).
[66] V. L.Deringer, R.Dronskowski, S.Maintz, A. L.Tchougréeff. LOBSTER: A tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem., 37, 1030-1035(2016).
[67] G.Henkelman, E.Sanville, W.Tang. A grid-based Bader analysis algorithm without lattice bias. J. Phys.: Condens. Matter, 21, 084204(2009).
[68] A. I.Boldyrev, B. D.Dunnington, T. R.Galeev, J. R.Schmidt. Solid state adaptive natural density partitioning: A tool for deciphering multi-center bonding in periodic systems. Phys. Chem. Chem. Phys., 15, 5022(2013).
[69] G. A.Olah, G.Rasul, G. K.Surya Prakash. N62+ and N42+ dications and their N12 and N10 azido derivatives: DFT/GIAO-MP2 theoretical studies. J. Am. Chem. Soc., 123, 3308-3310(2001).
[70] L. P.Cheng, Q. S.Li. Aromaticity of square planar N42− in the M2N4 (M = Li, Na, K, Rb, or Cs) species. J. Phys. Chem. A, 107, 2882-2889(2003).
[71] L. P.Cheng, Q. S.Li. N4 ring as a square planar ligand in novel MN4 species. J. Phys. Chem. A, 109, 3182-3186(2005).
[72] H. M.Buck, R. A. J.Janssen, G.van Zandwijk. 6π aromaticity in four-membered rings. J. Am. Chem. Soc., 112, 4155-4164(1990).
[73] L. P.Cheng, Q. S.Li. Theoretical study of nitrogen-rich BeN4 compounds. J. Phys. Chem. A, 108, 665-670(2004).
[74] X. F.Hao, X. J.Liu, J.Meng, Z. J.Wu, H. P.Xiang, E. J.Zhao. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B, 76, 054115(2007).
[75] M.Avalos Borja, M.Moreno Armenta, A.Reyes-Serrato.
[76] R. J.Needs, C. J.Pickard. High-pressure phases of nitrogen. Phys. Rev. Lett., 102, 125702(2009).
[77] C.Dickinson, M. J.Kamlet. Chemistry of detonations. III. Evaluation of the simplified calculational method for Chapman-Jouguet detonation pressures on the basis of available experimental information. J. Chem. Phys., 48, 43(1968).
[78] X.Li, H.Niu, A. R.Oganov, J.Zhang. Pressure-stabilized hafnium nitrides and their properties. Phys. Rev. B, 95, 020103(R)(2017).
[79] J. P.Agrawal. High Energy Materials: Propellants, Explosives and Pyritechnics(2010).
[80] G. T.Furukawa, M. L.Reilly. Heat capacity and thermodynamic properties of α-beryllium nitride, Be3N2, from 20 to 315 K. J. Res. Natl. Bur. Stand., Sect. A, 74A, 617-629(1970).
[81] T. B.Douglas, W. H.Payne. Measured enthalpy and derived thermodynamic properties of alpha beryllium nitride, Be3N2, from 273 to 1200 K. J. Res. Natl. Bur. Stand., Sect. A, 73A, 471-477(1969).
[82] G. E.Gurr, D.Hall, G. A.Jeffrey. Zur kenntnis des systems Be3N-Si3N4. V. A refinement of the crystal structure of β-beryllium nitride. Z. Anorg. Allg. Chem., 369, 108-112(1969).
[83] S.Chariton, N.Dubrovinskaia, L.Dubrovinsky, T.Fedotenko, D.Laniel, V.Milman, A.Pakhomova, V.Prakapenka, B.Winkler. High-pressure polymeric nitrogen allotrope with the black phosphorus structure. Phys. Rev. Lett., 124, 216001(2020).
[84] N. J.English, M.Li, C.Lin, X.Liu, H.-K.Mao, J. S.Smith, J. S.Tse, B.Wang, W.Yang, X.Yong. Temperature-dependent kinetic pathways featuring distinctive thermal-activation mechanisms in structural evolution of ice VII. Proc. Natl. Acad. Sci. U. S. A., 117, 15437-15442(2020).
[85] H.Dong, S.Li, X.Li, C.Lin, X.Liu, J. S.Smith, J. S.Tse, B.Wang, D.Yang, W.Yang. Temperature- and rate-dependent pathways in formation of metastable silicon phases under rapid decompression. Phys. Rev. Lett., 125, 155702(2020).


Set citation alerts for the article
Please enter your email address

© Copyright 2018-2021 | Chinese Laser Press. All Rights Reserved 沪ICP备15018463号-20