Jongchan Park, David J. Brady, Guoan Zheng, Lei Tian, Liang Gao. Review of bio-optical imaging systems with a high space-bandwidth product[J]. Advanced Photonics, 2021, 3(4): 044001
Copy Citation Text
Optical imaging has served as a primary method to collect information about biosystems across scales—from functionalities of tissues to morphological structures of cells and even at biomolecular levels. However, to adequately characterize a complex biosystem, an imaging system with a number of resolvable points, referred to as a space-bandwidth product (SBP), in excess of one billion is typically needed. Since a gigapixel-scale far exceeds the capacity of current optical imagers, compromises must be made to obtain either a low spatial resolution or a narrow field-of-view (FOV). The problem originates from constituent refractive optics—the larger the aperture, the more challenging the correction of lens aberrations. Therefore, it is impractical for a conventional optical imaging system to achieve an SBP over hundreds of millions. To address this unmet need, a variety of high-SBP imagers have emerged over the past decade, enabling an unprecedented resolution and FOV beyond the limit of conventional optics. We provide a comprehensive survey of high-SBP imaging techniques, exploring their underlying principles and applications in bioimaging.
Information requirements in bio-optical imaging are ever increasing. This demand is due to the landscape shift in contemporary biology, from morphological explorations and phenotypic probing of organisms, to an ongoing search for quantitative insights into underlying mechanisms at cellular and molecular levels. For example, observing large-scale neuronal activities of a brain1 requires an imaging system with subcellular resolution within a field-of-view (FOV) that encompasses the whole brain. To image a whole mouse brain of volume with resolution requires 500 billion spatial samplings, an enormous quantity that is far beyond the acquisition bandwidth of most current imaging systems.
For optical imaging, the information content is commonly described by the space-bandwidth product (SBP), a dimensionless quantity that equals the number of optically resolvable spots within an FOV.2,3 The higher the SBP, the more information we acquire, and the richer the measurement. In practice, the SBP of an imaging system is determined by two factors: the pixel count of the camera and the performance of optics. With recent advances in large-format image sensors, imaging optics have become the bottleneck in achieving a large SBP—in conventional imaging systems, the fundamental limit is optical diffraction while the practical limits are the geometrical aberrations and mechanical/thermal constraints of constituent components. For example, a high-performance objective lens (Olympus, UPlanSAPO20X) with magnification and a 0.75 numerical aperture (NA) has an FOV of 1.35 mm in diameter and captures a spatial frequency content up to at 550 nm. Neglecting the aberrations, a total SBP is . In practice, for state-of-the-art microscope objective lenses with similar form factors, a typical SBP varies from a few million to tens of millions. By contrast, current high-resolution complementary metal-oxide-semiconductor (CMOS) sensors can have as many as 250 million pixels.4 Even commercial smartphones with 100-megapixel cameras are available,5 far exceeding the SBP of conventional lenses.
To increase the SBP, the conventional approach relies on complicated lenses, resulting in a bulky setup and costly fabrication. Even with a long history of continuing effort, very few modern optical systems with a large aperture can achieve the diffraction-limited performance across a large FOV—we are approaching the end of the Moore’s law-like limit that the SBP of the system can be hardly improved solely by manipulating the lens parameters.6 To overcome this limitation, modern approaches utilize three strategies. The first strategy, referred to as the spatial-domain method, captures multiple images in the spatial domain to scale up the SBP. Representative techniques encompass array microscopy7–11 and multiscale optical imaging.12–14 The second strategy, referred to as the frequency-domain method, augments the SBP by performing a series of measurement in the Fourier domain. Within this category, the most important techniques include Fourier ptychography15–19 and structured illumination microscopy (intensity20,21 and complex field imaging22–25). It is worth noting that both spatial- and frequency-domain methods leverage the advantage of small-aperture optics in managing the lens aberrations.12 By contrast, the third strategy—wavefront-engineering-based methods—utilizes large-aperture lenses. The correction for the lens aberrations is accomplished by altering the phase of the wavefront through either the hardware-26–28 or computation-based approaches.29–32
Sign up for Advanced Photonics TOC. Get the latest issue of Advanced Photonics delivered right to you!Sign up now
In this review, we provide a comprehensive survey of these high-SBP imaging techniques in a unified framework, divulging their underlying principles, interconnections, and comparative advantages in bioimaging. We first introduce the concept of the SBP and discuss its relationship with the information capacity of an optical imaging system. The subsequent sections focus on the strategies to increase the SBP and their application in bioimaging. Finally, we summarize the field and provide perspectives.
The scope of this review is limited to the methods that increase the SBP of the imaging system rather than correcting for the sample-induced aberrations and scatterings.33–38 In practice, the lensless on-chip microscopy systems have been known for high-SBP imaging because of the absence of imaging lenses.39–43 However, they are only applicable to samples in proximity to the image sensor, restricting the breadth of biological applications.44,45 Therefore, we exclude them from the discussion herein.
2 Bioimaging and Space-Bandwidth Product
2.1 Limited Performance of Conventional Imaging Systems
Most bio-optical imaging systems are built upon refractive optics, where the light emanating from an object passes through a series of refractive lenses and forms an image on an image sensor. The paths of the light rays are mainly governed by the surface curvatures and refractive index of the constituting lenses, which bend the light rays following Snell’s law. Under the paraxial approximation, the light rays converge to a perfect focal spot. However, with an increased incident angle to the surface normal the paraxial approximation fails, and the light rays are refracted to a direction that deviates from the nominal focus. The aberrations so induced are functions of both the field height from the optical axis (or field angle) and aperture size. Therefore, the larger the FOV and aperture size (i.e., the larger the SBP), the worse the aberrations.
Correcting for aberrations in a system with a large FOV and aperture is a nontrivial problem. Conventional lens design techniques such as lens bending/splitting, stop shifting/symmetry, and use of aspherical surfaces often lead to a complicated configuration with tens or even hundreds of lenses, incurring a prohibitive fabrication cost and a large form factor. For instance, a high-performance photolithography objective lens supports a high NA ( in the air) across a large FOV ().46 However, the total length of stacked lenses is , and they weigh several hundred kilograms. Such a bulky and complex imaging system is unsuitable for use in bioimaging in a laboratory or clinical setting. Recently, McConnell et al.47,48 developed a microscope lens called the mesolens comprising 15 optical elements of up to 63 mm in diameter. The lens provides a 6-mm FOV and a 0.5 NA, enabling a resolution approximately four times higher than a state-of-the-art objective lens with a similar FOV. Despite being a significant advance, the FOV provided is still insufficient for large-scale bioimaging such as interrogating the functional connectivity of the brain in large animals.49 In this regard, this review excludes the imaging systems that improve the SBP purely through the conventional lens design process.
2.2 Space-Bandwidth Product
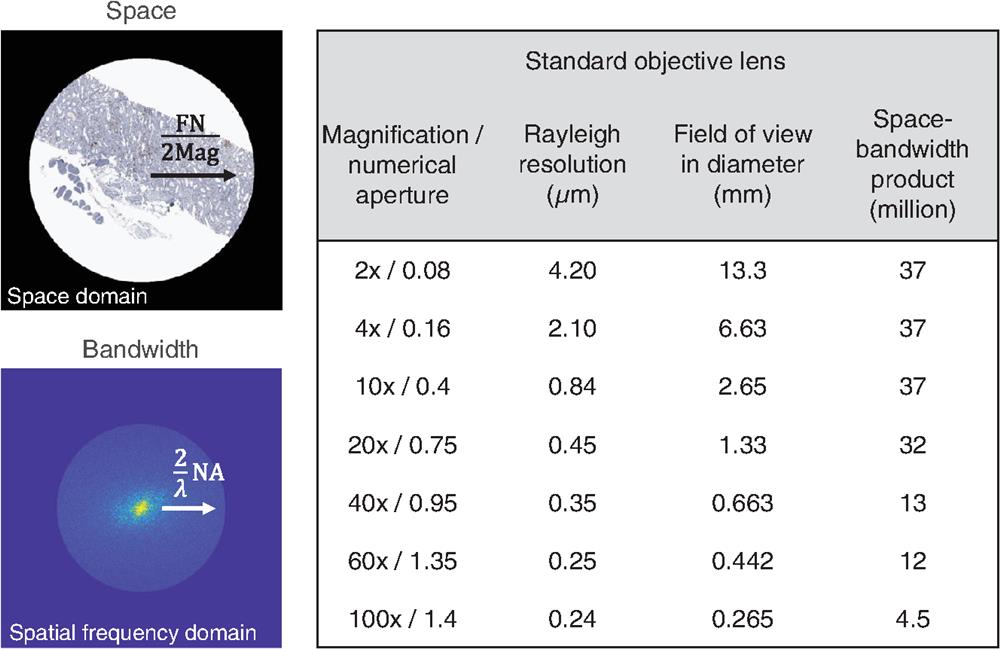
The SBP of an imaging system is a dimensionless number proportional to the information throughput, and it is usually calculated as the product of the FOV (space) and the spatial frequency range (bandwidth). The SBP is also referred to as Shannon number, the minimum number of samples required to completely determine the signal,50,51 or the maximum number of resolvable spots over the FOV. For example, for a given objective lens with a field number, FN, and a magnification, Mag, the FOV equals . In coherent imaging, the cut-off spatial frequency of the lens is for a complex amplitude, where NA and are the numerical aperture of the objective lens and wavelength, respectively. The cut-off frequency increases by a factor of two in incoherent imaging, with a triangular optical transfer function.51 For incoherent imaging, the diffraction-limited SBP equals the product of the FOV and spatial-frequency range: The FN is limited by the field diaphragm, which is bounded by the form factor of the objective lens. In general, a high-NA objective lens tends to have a relatively small SBP (Fig. 1), because the magnification of an objective lens does not increase at the same pace as that of its NA. For instance, at a 550 nm wavelength, the SBP of a objective lens (Olympus, UPlanSApo10X) is , and this number drops dramatically to for a objective lens (Olympus, UPlanSApo100X). Because of this nonlinear dependence, it is challenging to build an imaging system simultaneously with a high resolution and a large FOV.
Figure 1.The diffraction-limited SBP of standard microscope objective lenses at a 550 nm wavelength under incoherent illumination. The pathology slide image is modified from a public repository of image datasets (Image Data Resource).52,53
Although the SBP well quantifies the spatial degree of freedom of the optical system, the amount of information measured by the system must be discussed in conjunction with the signal-to-noise ratio (SNR). More specifically, the information capacity of each resolvable point under a white Gaussian noise is given as ,54 which monotonically increases with the SNR. Therefore, the total information capacity of an imaging system with two independent polarization states is55–57
It is worth noting that the information capacity does not directly state the Rayleigh resolution or FOV of the imaging system. Instead, it serves as the theoretical upper bound. For example, even with a high-resolution imaging system, a severe noise will deteriorate the image quality and, therefore, the practical resolution. The relationship between the information capacity and the SNR was first presented by Fellgett et al.55 Cox and Sheppard56 further discussed the information capacity in conjunction with the resolution. And later, this framework was extended to super-resolution microscopies, such as structured illumination microscopy and single-molecule localization microscopy.58,59
In a system with a nonuniform spatial resolution across the FOV, such as foveated lenses,60,61 the SBP is not equal to the product of the FOV and spatial frequency bandwidth. Instead, it must be calculated as a total number of spatially resolved spots. For simplicity, we confine the discussion to systems with a uniform spatial resolution.57
3 High SBP Imaging: Spatial-Domain Methods
3.1 Array Microscopy
A simple method to increase the SBP is to scan a sample using a high-NA objective lens and stitch the high-resolution images. However, because high-NA objective lenses normally have a small FOV (Fig. 1), the scanning of a single objective lens leads to a prolonged acquisition. For example, to scan a FOV with a objective lens, it takes , provided that the combined scanning and camera exposure time at each step is 1 s, which is typical for wide-field fluorescence imaging. This imposes a demanding requirement on the mechanical stability of the system. Also, an autofocusing system is required because the image can be easily defocused due to the misalignment between the scanning direction and sample plane or environmental variations, such as temperature fluctuation. Moreover, this acquisition scheme applies only to static samples. The motion of the object could, otherwise, introduce severe artifacts. Despite challenges described above, the step-and-repeat scanning method has achieved a remarkable success in whole-slide pathological imaging.62–65
To address these issues, Weinstein et al.7 developed a parallelization scheme using an array of microscopes [Figs. 2(a) and 2(b)]. Rather than using a single objective lens, they populated the objective lenses into an array, increasing the FOV by a factor of (number of objective lenses) while maintaining the high resolution of an individual lens.7 Because the FOV of each lens is small, the geometrical aberrations can be well corrected using relatively simple optics. The team used microlenses with a 0.65 NA and a 0.25-mm FOV. The correspondent SBP of an individual microlens is . The total SBP of the system in this acquisition scheme is scalable—it is proportional to the number of microlenses in the array. With a total of 80 microlenses, the system can capture an image with a total SBP of in a snapshot, surpassing the performance of the conventional objective lenses (Fig. 1). Using this system, the team demonstrated digital scanning of a large pathology slide across a area with a submicron spatial resolution [Fig. 2(c)]. The array of microscopes was initially developed for wide-field transmission imaging. Recently, this method has been extended to fluorescence imaging as well [Fig. 2(d)].9,67,68
Figure 2.Array microscopy. (a) Images are captured through parallelized microimaging systems. (b) Schematics of an array microscopy for digital histopathology. In their system, three lenslet arrays are stacked. Each lens group has a diameter of 1.5 mm and a working distance of . The microlenses are densely packed, and the orientation of the array is slightly tilted to the scanning axis. Therefore, a single-axis scan can provide the whole FOV. (c) Images of a pathology slide with a high SBP (). (d) Image of a fluorescently stained rat femur (upper) and its enlarged view (bottom) with parallelized scanning fluorescent microscopy. The scale bar for the top image is 1 mm, and the zoom-in images are . (e) Sequential illumination of the beam for mechanical scanning-free parallel imaging. Panels (b), (c), (d), and (e) are modified from Refs. 7, 9, and 66, respectively.
Despite parallel image acquisition, array microscopy still requires scanning to capture a complete picture of the sample. On the array, each objective lens forms a magnified image on the camera. To avoid the overlap between the adjacent images, the dimension of the magnified image cannot exceed the lens pitch, . Given a magnification , the maximum FOV of an individual lens at the object side is . Therefore, there is a gap between adjacent areas imaged. To fill this information, one must scan the sample across a distance of along both in-plane axes. Therefore, the higher the magnification , the longer the scanning range. Although this scanning range is much smaller than that required in the single lens-scanning-based approach, mechanically translating the sample is slow and prone to motion artifacts. To mitigate this problem, McCall et al.66 replaced mechanical scanning with temporal sequential imaging [Fig. 2(e)]. They built separate illumination for each microscope and lit only a subset at a time. Because the adjacent FOVs are not imaged simultaneously, they can overlap on the camera, alleviating the trade-off between the magnification and the total acquisition time.
3.2 Multiscale Optical Imaging
In conventional lens design, large-FOV imaging systems are particularly vulnerable to off-axis aberrations such as coma, astigmatism, and field curvature, which are functions of the field height from the optical axis. Among these aberrations, the field curvature is the toughest to correct for—it solely depends on the refractive indices and optical powers of lenses, and typical lens design techniques such as lens bending/splitting or stop symmetry/shifting are inapplicable.69 For a coaxial imaging system, a practical method to flatten the field curvature is to add a negative lens close to the image plane.70 However, the field flattening lens introduces an astigmatism that complicates the optical design. In addition, the imperfections of the lens surface such as scratches, dirt, and dust, would appear superimposed on the image.
A multiscale optical architecture addresses the field curvature issue by utilizing a large-scale main lens and a small-scale lenslet array [Fig. 3(a)]. The object is first imaged by the primary main lens onto a curved Petzval surface. This intermediate image is then relayed by the secondary lenslet array to an array of cameras on a curved surface. The resultant images are computationally combined to reproduce a large FOV. Because the field-dependent focal shift can be physically compensated for by moving individual cameras to the correspondent focal positions, this method possesses a key advantage that the field curvature can be loosely tolerated when designing the main lens, thereby easing the correction of other aberrations. Particularly, when imaging a distant object, the primary main lens can be a simple ball lens with an aperture at the centre [Fig. 3(b)].71 Because of the rotational symmetry about the chief rays, no off-axis aberrations are introduced. The resultant system, referred to as a monocentric camera,72–74 exhibits only spherical and chromatic aberrations, which can be further corrected for using multiple concentric layers of different refractive indices.75 However, due to the use of a large number of lenslets and cameras () at the focal surface, the early monocentric cameras are generally bulky. For example, the first-generation monocentric gigapixel imaging system with 98 cameras13 has a volume of and a weight of 93 kg. To improve the form factor, Karbasi et al.76 replaced the lenslet array using a curved fiber bundle, directly transmitting the focal surface image to a single large-format camera. The resultant 25-megapixel monocentric imaging system has a footprint of . Kim et al.77 recently developed a more compact system by placing a bio-inspired hemispherical silicon nanorod photodiode array at the focal surface of the ball lens.
Figure 3.Multiscale optical systems. (a) Illustration of multiscale optical designs. (b) Schematic of the AWARE-2 camera consisting of multiscale optics and 98 microcameras. (c) The camera captures a 0.96 gigapixel image. (d) Multiscale optical system for bioimaging. The system can track traces of GFP-labeled immune cells. The scale bars are 1000 and . Panels (b)–(d) are modified from Refs. 13, 14, and 71, respectively.
Both multiscale optical imaging and array microscopy78 leverage the same fact that smaller lenses outperform larger lenses in image quality. Here, the performance of a lens is quantified as the ratio of SBP achieved to the theoretical maximum for a given magnification and FOV. While the multiscale lens system generally includes a hierarchy of aperture sizes stepping the field down from the primary lens to small-scale lenslets, the array microscopy consists of only one-level structure. In microscopy, the benefit of using a primary lens is that it can premagnify the image for the secondary lenslets to process. Therefore, the magnification of the secondary lenslet can be less than one, allowing the individual FOVs at the focal surface to be overlapped. The complete picture of the sample can be captured in a snapshot without scanning. Also, the use of the primary lens allows a large standoff, which is critical for imaging distant scenes. However, the downside is the introduction of additional aberrations by the primary lens. Therefore, the secondary lenslets must correct for these aberrations in addition to relaying the image.
Using a multiscale microscopy system, Fan et al.14 demonstrated video-rate imaging of biological dynamics at centimetre scale and micrometre resolution [Fig. 3(c)]. A custom primary objective lens with a working distance of 20 mm images a large-FOV fluorescence scene. The intermediate focal image is segmented and relayed by the secondary lenses arranged on a curved surface. The final individual images are measured by an array of sCMOS sensors. Using this system, the team demonstrated calcium imaging of the nonuniform propagation of epileptiform neural activities.
4 High SBP Imaging: Frequency-Domain Methods
In contrast to array microscopy/multiscale optical systems where the individual images are stitched in the spatial domain, the frequency-domain methods combine the images in the spatial-frequency domain (Fourier domain). Because a translational shift in the Fourier domain corresponds to an angular shift in the real space,51 the images associated with various spatial-frequency components can be measured by illuminating the sample at varied angles or patterns, eliminating the need for mechanical scanning. Within this category, representative techniques encompass Fourier ptychography and structured illumination microscopy.
4.1 Fourier Ptychography
Fourier ptychography15 is a computational imaging technique that can capture high-SBP images using low-cost, small-aperture imaging systems. By varying the illumination angle, Fourier ptychography shifts the frequencies of the object information in the Fourier domain followed by passing the components that fall within the aperture of the imaging system. The images so obtained are subaperture representations of the object, and they can be computationally combined in the Fourier domain to compose a large aperture (Fig. 4). Zheng et al. first demonstrated this method in optical microscopy and reported an SBP of 0.23 billion for a complex amplitude image.15,81 The state-of-the-art implementation achieved 1.45 NA using a objective lens in the air [Fig. 4(d)].79 An oil-immersion condenser lens can also be used with a objective to achieve an NA of 1.6.19 The resultant SBP is two orders of magnitude higher than that of a benchmark objective lens (Olympus ).
Figure 4.High-SBP imaging with Fourier ptychography. (a) Principles of spatial frequency-domain multiplexing. (b) Simplified diagram of a phase-retrieval algorithm. (c) Recovery of the spatially varying pupil function. (d) High-resolution Fourier ptychography image of red blood cells. Particles are shown in the zoom-in view of malaria-infected red blood cells (arrow). Panels (c) and (d) are modified from Refs. 79 and 80, respectively.
We illustrate the operating principle of Fourier ptychography in Fig. 4(a). Under the on-axis illumination, the addressable spatial frequency range of the system is an area of a circle with a diameter of in the Fourier domain (coherent transfer function). Here, is the wavelength, and , where and are the entrance pupil diameter and focal length of the small-aperture lens, respectively. The center of the circle coincides with the origin of the Fourier space. Under angled illumination, the chief ray of the diffraction light cone changes with the illumination, leading to a linear shift of the frequency representation in the Fourier domain. The shifted distance equals , where is the incident angle of illumination. Therefore, the high-frequency components of the sample that is initially blocked by the aperture of the imaging system can be collected. By capturing a series of images under varied illumination angles and stitching their frequency representations in the Fourier domain, we can recover a large spatial frequency range of the object.
The name of Fourier ptychography comes from a related lensless imaging modality, ptychography.82 With ptychography, the object is typically illuminated by a spatially confined beam at the spatial domain. The far-field diffraction patterns are then recorded at the spatial frequency domain as the object is mechanically scanned to different positions.83 Fourier ptychography swaps the spatial domain and the Fourier domain via a lens. With Fourier ptychography, the confined support constraint is imposed by the pupil aperture in the spatial frequency domain while the images are recorded in the spatial domain. In contrast to the mechanical scanning process in ptychography, Fourier ptychography scans the object’s Fourier spectrum in the spatial frequency domain via angle-varied illuminations. Fourier ptychography also shares its root with synthetic aperture imaging, which was first developed in radio astronomy for bypassing the resolution limit of a single radio telescope.84 A similar concept has been demonstrated for light microscopy where intensity and phase information are measured via interferometric setups.22,85–88
With Fourier ptychography, however, no direct phase measurement is needed in the acquisition process. Instead, the phase information is recovered from the intensity images using an iterative process referred to as phase retrieval.89–91 One widely adopted algorithm for phase retrieval is alternating projection,92 which iteratively imposes object constraints in the spatial and Fourier domains. For Fourier ptychography, the measured intensity is used as a modulus constraint in the spatial domain, and the confined pupil aperture is used as a support constraint in the Fourier domain [Fig. 4(b)].93
The nonreliance on direct phase measurement in Fourier ptychography eliminates the challenges of interferometry-based techniques, such as inherent speckle noise and sensitivity to phase errors. In addition, a Fourier ptychography microscope can be built with low-cost optics,94 facilitating its use in point-of-care applications.95–98 On the other hand, since the phase information cannot be directly measured as in interferometry, the recovery of a complex amplitude from intensity-only images is computationally expensive. This drawback can be alleviated using parallel processing via a graphic processing unit or by machine-learning-related approaches.99–101
To reconstruct a high-fidelity phase map, Fourier ptychography requires data redundancy in the Fourier domain.102 The Fourier spectrum of the measured image must be overlapped with the adjacent measurement—each data point in the Fourier domain needs to be included in at least two measurements to avoid ambiguity in the phase-retrieval process,103 a fact that substantially increases the data acquisition time. In addition, the small collection aperture of the lens limits the range of the measurable Fourier spectrum, leading to a reduced signal level under dark-field illumination. A long exposure time is thus required to capture images with a high SNR. To alleviate this problem, Tian et al.17 developed a multiplexed illumination strategy that illuminates the sample with beams at multiple, randomly selected incident angles.17 They demonstrated that the total number of images can be significantly reduced without sacrificing the reconstructed image quality. Alternatively, nonuniform Fourier sampling104 and data-driven approaches can be employed to reduce the number of image acquisitions.99,100,105
The measurement of a complex-amplitude image in Fourier ptychography enables great flexibility for postacquisition processing. For example, both the aberrations of the objective lens [Fig. 4(c)]15,80,106–111 and the defocus of the sample112,113 can be numerically corrected for, even under severe conditions.94,114 Based on this principle, Chung et al.115 reported a Fourier ptychographic retinal imaging method that can correct for eye lens aberrations and thereby enable full-resolution imaging of the retina. Similarly, postacquisition digital refocusing can be used to extend the depth of field for imaging microfilters containing captured tumor cells,97 96-well plate,112 blood smear,109 and pathological slides.113
One major limitation of Fourier ptychography is its reliance on angled illumination—for a three-dimensional (3D) object, tilting the illumination would change the object’s spectrum rather than just shifting it in the Fourier domain. As such, Fourier ptychography has been primarily used in imaging optically thin samples in transmission mode. To handle 3D thick specimens, it is possible to employ fixed illumination and modulate the light waves in the detection path.16,116 In this case, the recovered image represents the exiting wavefront of the object, which can then be digitally propagated back to any plane along the optical axis. The object thickness becomes irrelevant in the modeling. Also, recent advances in light scattering models have enabled reflection-mode Fourier ptychography,117,118 which can be further integrated with the modulation concept for deep tissue imaging.111 It is worth noting that Fourier ptychography is inapplicable to fluorescent samples because the fluorescence emission is generally isotropic and independent of illumination angles.119,120
4.2 Structured Illumination Microscopy
Structured illumination microscopy20,121,122 is also a frequency-domain method based on incoherent imaging. However, unlike Fourier ptychography, structured illumination microscopy shifts the frequency representation of an object through patterned illumination,20,122,123 making it suitable for fluorescence imaging. In a typical setup, the sample is illuminated by a striped pattern of a specific frequency, . The resultant image is the product of the object function and the illumination pattern. The frequency shifting property of the Fourier transform implies that the frequency representation of the image is shifted by with respect to the original spectrum. Therefore, the high frequencies beyond the aperture of the imaging system can be collected [Fig. 5(a)]. The structured illumination microscopy is primarily implemented in the epi-illumination mode. Because the illumination and imaging paths share the same objective lens, the upper illumination pattern frequency is bounded by the bandwidth of the objective lens for linear imaging, leading to a maximal resolution improvement.
Figure 5.Structured illumination microscopy. (a) Fourier domain representation of conventional, linear, and nonlinear structured illumination microscopy. In conventional microscopy, the measurable spatial frequency range is given as . In linear structured illumination, spatial frequency information of the sample is laterally shifted an amount corresponding to the period of the illuminating pattern. Therefore, the high-spatial frequencies beyond the conventional imaging system become observable. In nonlinear structured illumination, the spatial frequency information of the sample is shifted corresponding to integer multiples of the pattern’s frequency. With pattern rotation, a large spatial frequency range can be collected. (b) A mammalian CHO cell imaged by the nonlinear structured illumination microscopy. Panel (b) is modified from Ref. 124.
With nonlinear excitation21 or plasmonic substrates,125 the resolution can be further enhanced. In this case, the image is the product of the object function and the illumination function to the power of , where , and it describes the nonlinear dependence of the emitted light on illumination. Again, based on the frequency shifting property, the frequency representation of the object is shifted by , thereby increasing the observable frequency radial range by a factor of . By rotating the illumination pattern and varying its frequency, one can record all the frequency components of the object within an area of , expanding the SBP of the objective lens by a factor of . Using this strategy, Rego et al.124 demonstrated a 50-nm resolution within an FOV of [Fig. 5(b)]. It is worth noting that the resolution of structured illumination microscopy cannot be increased arbitrarily. Instead, given a limited photon budget of a fluorescent sample, an achievable resolution is limited by SNR.58
Based on the principle of structured illumination microscopy, various super-resolution imaging systems have been developed to increase the spatial resolution without sacrificing the FOV. For example, scanning structured illumination microscopy126 increases the spatial resolution of laser scanning microscopy with patterned illumination or detection.127–132 Image scanning microscopy133–135 increases the resolution of confocal microscopy136–138 by a factor of simply by replacing the point detector of the confocal microscope with an array detector. Moreover, these super-resolution scanning microscopy methods provide optical sectioning, thereby enabling imaging of relatively thick biological samples. It is worth noting that structured illumination microscopy has been traditionally used as a super-resolution imaging technique. Further increasing the resolution and optical sectioning ability is an important direction. However, imaging across a large FOV has not been pursued actively, and only recently has its potential as a high SBP imager with millimeter-scale FOV been discussed.139
5 High SBP Imaging: Wavefront-Engineering-Based Methods
Both spatial- and frequency-domain methods leverage the advantage of small-aperture optics in managing the lens aberrations. By contrast, wavefront-engineering-based methods utilize large-aperture lenses. The correction for the lens aberrations is accomplished by wavefront modulation through either hardware or computation.
5.1 Hardware Approaches
To modulate the wavefront, the hardware approaches use devices such as a deformable mirror140,141 or a liquid-crystal spatial light modulator (SLM).142 Because aberrations depend on the field height, the corresponding distorted wavefronts must be corrected for at individual field points/areas. Therefore, the hardware approaches are commonly implemented in scanning-based systems, sequentially acquiring image patches in which the aberrations can be considered homogeneous.
Within this category, the most important method is adaptive scanning optical microscopy26,143,144[Fig. 6(a)]. A custom large-aperture objective lens collects light emitted from an object. A galvanometric scanning mirror is placed at the back aperture of the object lens. Because the chief rays associated with different field heights are incident on the galvanometric scanning mirror at varied angles, scanning these rays in the angular domain passes the correspondent field areas to the following imaging optics in a sequential manner. A deformable mirror is placed at a conjugated pupil plane, adding the precalibrated phase delays to the wavefront and thereby compensating for the aberrations at the field location scanned [Fig. 6(b)]. The use of adaptive optics releases the design constraint on the large-aperture objective lens because the deformable mirror readily compensates for low-order wavefront distortions characterized by Zernike modes.145,146 Using this system, the team designed a system with a resolution (0.21 NA at 510 nm wavelength) across an FOV of , leading to an SBP of 2.7 billion. The system is highly stable because there is no mechanical translation of the sample or imaging optics. In addition, the system can image selected sub-FOVs at a high-frame rate, instead of imaging the entire FOV. Using this strategy, Potsaid et al. demonstrated real-time imaging of multiple live C. elegans worms [Fig. 6(c)].143 Although not being demonstrated by the authors, the system can scan the image in the axial direction by superimposing a quadratic phase map on the wavefront using a high-resolution SLM.
Figure 6.Hardware wavefront-engineering-based methods for high-SBP imaging. (a) System schematic of adaptive optical scanning microscopy. (b) The viewing location is given by the tilting angle of the galvanometric mirror, and the corresponding aberrations are corrected by the deformable mirror. (c) A bright-field image of a living C. elegans in a sub-FOV of the system. (d) Principles of high-resolution wide-FOV focusing with a disordered metasurface and wavefront shaping. (e) Scanning fluorescence microscopy with the metasurface. Immunofluorescence-labeled parasites (Giardia lamblia cysts) were imaged. The FOV and resolution are comparable to that of the objective lens and objective lens, respectively. Panels (a), (b)–(c), (d)–(e) are modified from Refs. 27, 143, and 144, respectively.
Adaptive scanning optical microscopy requires the precalibration of the system at each field location. Therefore, the target of interest must be directly accessible to the microscope. If there is a layer of substance with unknown aberrations between the target and the microscope, such as a coverslip, an immersion medium, or a heterogeneous structure, this method will fail to acquire the aberration-free images. To correct for the sample-induced aberrations, the system must also use a wavefront sensor, such as a Shack–Hartmann sensor, to measure the wavefront aberration, followed by correction using the wavefront modulator. The resultant systems are particularly useful for quasiballistic imaging of volumetric samples. For example, adaptive optics optical coherence tomography has been demonstrated in retinal imaging, providing a single-cell resolution across different retinal layers.147–149 The correction of sample aberrations in such systems not only increases the resolution but also improves the SNR and thereby the penetration depth.150,151 A more specific article about this topic can be found elsewhere.35
The current hardware approaches cannot compensate for high-order wavefront distortions beyond the pixel count of the SLM. Also, the compensation pattern is bandlimited by the finite pixel pitch of the SLM. Therefore, in a highly aberrated imaging system, the imaging resolution is often lower than the diffraction limit even after wavefront correction. To solve this problem, Jang et al.27 demonstrated a wavefront-engineering system using a disorder-engineered metasurface and an SLM [Figs. 6(d) and 6(e)]. The metasurface consists of a subwavelength array of nanopillars with various widths that scatter light at very large angles up to 0.9 NA. By controlling the incident wavefront on the metasurface through the SLM, the team fully utilized the large scattering angle of the metasurface for tight focusing across a large FOV. This is equivalent to using a wavefront modulator with a reduced pixel size and an increased pixel count at the expense of decreased contrast. Using this system, the team demonstrated high-resolution () large-FOV (8 mm in diameter) scanning fluorescence microscopy. The corresponding SBP is 0.22 billion, which is much greater than those of conventional objective lenses. However, the phase map on the SLM must be updated sequentially during scanning, leading to a slow acquisition speed.
5.2 Computational Approaches
With recent progress on computational optics, the geometrical aberrations can also be numerically corrected for in postprocessing, increasing the SBP of the system at a given geometry. Because this approach does not increase the hardware complexity, it can be readily implemented in off-the-shelf imaging systems.
Based on Fourier optics principles, the complex generalized pupil function is proportional to the scaled optical transfer function, which is related to the point-spread-function (PSF) through the Fourier transform. Aberrations thus can be described as a phase term inside the generalized pupil function in a single-pass system. Given a unit magnification, the image, , is a convolution of the object function, , with the system’s aberrated PSF : where is the noise term. Transforming Eq. (3) to the Fourier domain gives where and are axes in the spatial frequency domain. , , , and are the Fourier transforms of , , , and , respectively. Because the system’s field-dependent can be measured as a prior, in the frequency domain, the image can be estimated as The propagation of the noise is determined by the condition number of , and the solution of Eq. (5) is well-posed only when .152
For coherent imaging, the coherent transfer function is complex, and it is described as Aberrations of the system will change only the phase term . Because 152 within the objective’s bandwidth, its effect on the image can be readily reversed by multiplying with the complex conjugate of . The complex coherent transfer function can be measured using an interferometric setup. An example is shown in Fig. 7(a).30
Figure 7.Computational wavefront-engineering-based methods for high-SBP imaging. (a) Computational correction of aberrations in optical coherence tomography and interferometric synthetic aperture microscopy. (b) Computational correction of spatially varying aberrations of a wide-FOV objective lens (). The system shows diffraction-limited performance over the entire FOV (13 mm in diameter). (c) Correcting spatially varying aberrations. The hardware approach sequentially corrects for aberrations at local positions. Correction of the whole FOV with averaged aberrations results in a degraded performance. By contrast, the computational approach can correct for spatially varying aberrations across the whole FOV without lateral scanning. Panels (a) and (b) are modified from Refs. 30 and 31, respectively.
For incoherent imaging, the numerical correction of the aberrated pupil function is nontrivial. In this case, the incoherent PSF equals The corresponding incoherent optical transfer function is the normalized autocorrelation of its coherent counterpart:153Given a circular aperture, has twice the bandwidth of , and its modulus monotonically decreases within this range, leading to a large condition number. Therefore, the solution of Eq. (8) is ill-posed.
To deconvolve the incoherent PSF, conventional methods use regularization or statistical algorithms.152 However, the results are sensitive to noise, and the improvement in resolution is often limited. To overcome these problems, Zheng et al.31,80 developed a multiplane method. Rather than capturing only one in-focus image, they captured multiple defocused images at varying depths, followed by retrieving the phase with an iterative algorithm.154 Using this method, the team demonstrated the computational correction of spatially varying aberrations of an objective lens (, Olympus) in a large FOV (13 mm in diameter). They recovered the aberrated pupil functions at 350 field locations and numerically remedied the associated wavefront distortions, leading to a diffraction-limited resolution within the entire FOV [Fig. 7(b)].
In general, the acquisition speed of computational wavefront engineering is faster than that of the hardware-based approach. In the computational approach, one can calculate the aberrated pupil function and perform corrections in postprocessing [Fig. 7(c)] by simply dividing the FOV into smaller segments in which the aberrations can be considered homogenous. By contrast, the hardware approach requires scanning and updating of the phase pattern on the wavefront modulator during the measurement, resulting in a slow acquisition.
6 Comparative Advantages
In this review, we categorize high-SBP bioimagers into spatial-domain methods, frequency-domain methods, and wavefront-engineering-based methods (Fig. 8). We reviewed representative works in each category and compared their achievable SBP in Fig. 9. Here, the SBP of the state-of-the-art microscope objectives serves as the baseline (dashed curve), representing the limit that conventional optics can reach. All modalities marked on this graph surpass this baseline, pushing the SBP limit toward the giga scale (dot-dashed line). To compare these methods, we use the spatial resolution (i.e., reciprocal of bandwidth), FOV, and temporal resolution as the metrics.
Figure 8.Illustration of various high-SBP imaging techniques. The pathology slide image is modified from a public repository of image datasets (Image Data Resource).52,53
Figure 9.SBP of high-SBP imaging systems. We note that the cut-off spatial frequency of an incoherent imaging system is double that of the coherent imaging system given the same NA. All frequency-domain methods15,19,22,24 are coherent imaging methods. In this graph, the SBP values of objective lenses were calculated for incoherent imaging. For coherent imaging, the cut-off spatial frequency of the objective lenses will be halved. The number in the “Ref” column next to the author and year indicates the corresponding reference index.
So far, spatial-domain methods9,10,14 and wavefront-engineering-based methods26–28,31 have been mainly used to expand the FOV with a moderate spatial resolution. It is challenging for these two categories of techniques to reach a resolution comparable to that of high-NA objective lenses (, , and ) in Fig. 9. For array microscopy, although we can replace each lens with a high NA objective, the practical hindrance lies in the trade-off between the NA and the depth of focus of individual lenses—the higher the NA, the smaller the depth of focus, the more sensitive the instrument to misalignment. For multiscale optical systems, the NA of the primary lens at the object side is proportional to its NA at the image side, which must in turn match with that of the secondary lenses. To increase the NA of the primary lens at the object side, we must increase the apertures of the secondary lenses as well, a fact that diminishes the advantage of using small aperture lenses in correcting for aberrations. Although we can use multilevel structures to further step down the aperture size, it increases the system complexity. For wavefront-engineering-based methods, with a given number of degrees of freedom to modulate the wavefront, the higher the NA, the lower the phase sampling density at the pupil plane. Therefore, high-order wavefront distortions beyond the degree of freedom of the wavefront modulator cannot be corrected for, resulting in a degraded resolution.
In frequency-domain methods, Fourier ptychography and structured illumination microscopy provide complementary capabilities of pushing the FOV and resolution beyond the limit of conventional optics. On the one hand, Fourier ptychography has been primarily used to image a large FOV where a small aperture lens collects the light. The frequency bandwidth is limited by the maximum illumination angle, which is in the air. Therefore, the maximum collection NA is less than one without an oil/water-immersion condenser.19 On the other hand, structured illumination microscopy has been predominantly used to boost the resolution of high-NA lenses, such as oil/water immersion microscope objectives, doubling their effective NA in linear imaging. However, when being applied to large-FOV imaging with a small aperture lens, it is not as effective as Fourier ptychography in expanding the frequency bandwidth because the maximum frequency of the illumination pattern in the linear scheme is limited by the lens’ small NA.
To compare the temporal resolution, we define a snapshot factor, , as the ratio of the SBP that is seen by the instrument at a time to the complete measurable SBP. A larger indicates a more time-efficient measurement and thereby a higher temporal resolution. For array microscopy, equals the FOV of an individual lens divided by its geometrical size. Given the FN, lens pitch , and magnification , the snapshot factor is . The geometrical factor of given by the circular FOV of the lens is neglected for simplicity. Therefore, the larger the , the smaller the , the lower the temporal resolution. For multiscale optical systems, , and the entire SBP can be acquired in a snapshot. For Fourier ptychography, given the bandwidth of a collecting objective, the number of required illumination angles is , where is the target frequency bandwidth, and the factor two is due to the oversampling requirement for phase recovery. Since the scanning is performed in the frequency domain, equals the inverse of , i.e., . Therefore, the larger the , the smaller the , the lower the temporal resolution. It is worth noting that the trade-off between and can be alleviated by employing multiplexed illumination.17 For structured illumination microscopy, has the same form as that in Fourier ptychography. For hardware wavefront-engineering-based approaches, is inversely proportional to the number of image patches in which the aberrations can be corrected for using a single-phase pattern displayed on the wavefront modulator. We summarize the comparative advantages discussed in Table 1.
Strategy
Imaging modality
Spatial resolution
Field of view
Temporal resolution (characterized by the snapshot factor )
Spatial-domain methods
Array microscopy
Moderate
Large
FN, field number; , magnification; , lens pitch.
Multiscale optical imaging
Moderate
Large
Frequency-domain methods
Fourier ptychography
Moderate
Large
, frequency bandwidth of the collecting objective; , target frequency bandwidth.
Structured illumination microscopy
High
Medium
, frequency bandwidth of the collecting objective; , target frequency bandwidth.
Wavefront-engineering-based methods
Hardware approach
Moderate
Large
, number of image patches
Computational approach
Moderate
N/A
N/A
Table 1. Comparative advantages of high-SBP imaging techniques. The FOV and temporal resolution of computational wavefront-engineering-based methods vary with the imager used. Therefore, we did not make conclusive comments.
The snapshot factor quantitatively describes how fast a system can image. The imaging speed of a snapshot system () is limited by only the readout speed of the camera, making it suitable for dynamic imaging of live biosamples. By contrast, the time-sequential methods () generally take larger-sized images at the expense of a reduced temporal resolution. Therefore, they are more suitable for imaging fixed specimens. The SNR of a system is also closely related to the snapshot factor, . Given the same number of exposures, the higher the snapshot factor, the higher the SNR. For example, when imaging a scene at a given frame rate, the SNR of a snapshot imager is times higher than that of scanning-based systems with .
Noteworthily, although we divide the high-SBP imagers into three categories, techniques are not mutually exclusive. There is an interesting trend to build hybrid imagers that cross these barriers. For example, Chan et al.112 developed parallel Fourier ptychography microscopy by combining the array of microscopes with Fourier ptychography. The optical system consists of 96 microscopy units. They improved the NA of each unit from 0.23 to 0.3 through Fourier ptychography. The team demonstrated this system in imaging a 96-well plate with an extended depth of field at 0.7 frames per second. As another example, the wavefront-engineering-based methods can be combined with array microscopy or multiscale optical imaging to reduce the residual aberrations and thereby further improve the resolution. We envision that an ideal high-SBP imager probably combines various techniques in a single device.
7 Outlook
7.1 Toward High Speed
The traditional definition of SBP does not account for the temporal dimension. For bioimaging, the ability to observe fast dynamics is as critical as having a large FOV and a high resolution, particularly for in vivo or live-cell imaging applications.155–157 In this review, we characterized the temporal resolution using a snapshot factor, . To incorporate the time dimension, it is rationale to revise SBP as space-bandwidth- product,158 which quantifies the information flux. For scanning-based high-SBP imagers, the acquisition of abundant space-bandwidth information usually comes at the expense of a reduced , as shown in Table. 1. In contrast, multiscale optical imaging offers the snapshot advantage, . A large snapshot factor is crucial for high-speed bioimaging because the SNR decreases with the frame rate at a given photon flux. For example, using a snapshot multiscale microscope for the first time, Fan et al. demonstrated cortex-wide structural and functional calcium imaging at a video rate (30 fps).14 The high space-bandwidth- product image provides valuable information about the long-range connectivity of neurons across the whole brain. However, the temporal resolution is still insufficient to image the propagation of cellular action potential—the fundamental phenomenon of transmitting information through neural networks, which rises and decays within milliseconds.159 In their system, further improving the frame rate is limited by the electronic data transfer rate from cameras to the host computers. Using burst imaging and storing the images on the camera board can potentially boost the frame rate up to several thousand frames per second, though the synchronization among cameras will be challenging.
For high space-bandwidth- product imaging, optimizing the data processing pipeline is equally important as acquisition.78 Given an enormous information flux, the extraction of the useful bio-information and relating it to cell/tissue physiology require new computational tools, such as multidimensional image analysis.160,161 The insights so obtained can potentially address fundamental questions such as how sensory inputs are dynamically mapped onto the functional activity of neural populations and how their processing leads to cognitive functions and behavior. Despite the substantial amount of studies, the exact mechanisms still remain elusive.162,163
7.2 Toward Super-Resolution
So far, most high-SBP imaging has been performed at scales from microscopic to macroscopic, with a resolution being fundamentally limited by diffraction. An imaging system that pushes the resolution toward the subdiffraction limit while maintaining a large FOV will serve as a vital tool to explore the connection between the molecular building blocks and overall tissue/cell functionalities. For example, large-FOV super-resolution imaging is instrumental to the study that relates assembly and disassembly of intracellular actin filaments with macroscopic behavior of complex biosystems or tissues.164 As another example, individual protein folding generally occurs on nanoscopic scales, but its energy landscape is modulated by myriad interactions at the whole-cell level.165 Large-FOV super-resolution imaging will be the enabling tool to reveal the spatial pattern of folding/unfolding in response to various cellular effectors, such as cellular water and transport machinery.166
As shown in Table 1, current high-SBP imagers face challenges in this realm. Only a structured illumination microscopy approach can provide such a high resolution but within a moderate FOV. A possible solution to implement high-SBP imaging is expansion microscopy,167–169 a technique that physically enlarges the sample in each dimension by chemical approaches, thereby unravelling the nanoscale information. At this expanded scale, the large-FOV imagers such as the microscope array, multiscale optical systems, and Fourier ptychography become applicable.
7.3 Toward 3D Imaging
Currently, high-SBP imagers have been mainly used for two-dimensional (2D) planar imaging. However, because most biosystems possess 3D structures, directly applying the high-SBP techniques to optically thick samples will lead to a reduced contrast and resolution. Therefore, implementing high-SBP imaging in 3D microscopy represents a cutting-edge direction. For 3D imaging, we can still use the conventional definition of SBP in 2D [Eq. (1)] but must associate it with a specific depth plane. Among all modalities discussed, only frequency-domain methods have been exploited for 3D imaging. For example, 3D Fourier ptychography has been demonstrated based on single-scattering models.170,171 However, these methods work for only optically thin samples, in which the first Born or the first Rytov approximation is valid.172 For optically thick samples, multiple light scattering makes it challenging to solve the associated 3D inverse problem, resulting in inaccurate reconstruction as well as a missing cone issue—the inaccessibility of central low spatial frequency information in the 3D Fourier spectrum along the optical axis of the imaging system.173 The multislice beam propagation model has emerged to be a promising computational technique for imaging highly scattering biological samples.174–178 Alternatively, structured illumination microscopy has been long used for 3D imaging of biostructures.179 Nonetheless, it suffers from an amplified noise attributed by the out-of-focus light,180 which reduces achievable resolutions.58
To expand the arsenal of high-SBP imaging tools applicable to 3D microscopy, one promising direction is to combine existing planar high-SBP techniques with light-sheet microscopy.181–183 The superior optical sectioning capability of light-sheet microscopy enables imaging of thick and inhomogeneous samples. For example, Liu et al.38 reported a high-SBP, 3D recording of a live zebrafish embryo by integrating light-sheet microscopy with adaptive optics. Yet, the FOV is limited by the detecting objective lens even with galvanometric scanning. With this regard, the integration of a multiscale microscopy system with light-sheet illumination will offer a significantly expanded FOV. Also, the large light collection efficiency of multiscale microscopy () will enable high-speed scanning of 3D samples, though a degraded resolution due to sample-induced aberrations is expected. Tailoring a large-sized light-sheet excitation beam is another issue; this may be accomplished with large-scale metasurfaces184 or wavefront shaping systems.27,185,186 Also, handling of extremely large 3D or four-dimensional data will be challenging.187
Jongchan Park, David J. Brady, Guoan Zheng, Lei Tian, Liang Gao. Review of bio-optical imaging systems with a high space-bandwidth product[J]. Advanced Photonics, 2021, 3(4): 044001