
Xiao-na Lia, Li-xue Jiang, Qing-yu Liu, Yi Ren, Gong-ping Wei. Hydrogen-Assisted C-C Coupling on Reaction of CuC3H- Cluster Anion with CO†[J]. Chinese Journal of Chemical Physics, 2020, 33(5): 628
- Chinese Journal of Chemical Physics
- Vol. 33, Issue 5, 628 (2020)
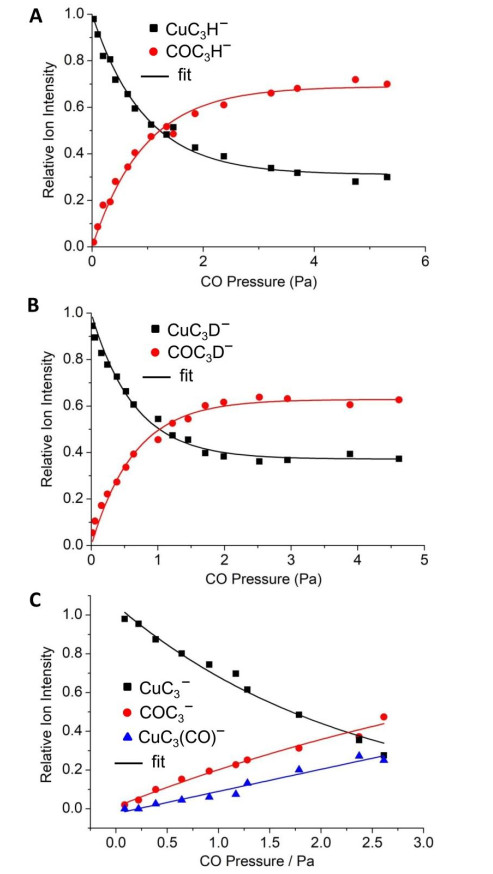
Fig. 1. Variation of ion intensity with respect to the pressures of CO on the reactions of CuC3H−
(a), CuC3D− (b), and CuC3− (c) with CO. The data points were experimentally measured, and the solid lines were fitted to the experimental data points on the basis of least-square procedure. The fitted results demonstrated that about 31% of laser-ablation generated CuC3H− and about 37% of such generated CuC3D− were inert toward CO.
Fig. 1. Time-of-flight mass spectra for the reactions of mass-selected CuC3H
Fig. 2. (A) The DFT-calculated low-lying isomers for CuC3H
Fig. 2. The density functional theory (DFT) calculated structures and relative energies of the CuC3H− and CuC3− isomers as well as the structures of products COC3H− and COC3−. The relative energies are in unit of eV and bond lengths are given in pm. Single point energies calculated at the CCSD(T) level are listed in the square brackets.
Fig. 3. Photoelectron spectra of CuC3H
Fig. 3. The DFT-calculated profiles on the transformation of different CuC3H− isomers shown in FIG. S2. The relative energies are in unit of eV and bond lengths are shown in pm. The superscripts "2" and "4" represent the doublet and the quartet electronic states, respectively.
Fig. 4. The DFT-calculated potential energy profiles for reactions (A) CuC3H
Fig. 4. The DFT calculated potential energy profile for reaction CuC3H− + CO on the doublet state. The relative energies are in unit of eV. The structures of intermediates and transition states are plotted. Bond lengths are given in pm.
Fig. 5. Natural charge in e distributions on I3, I5,
Fig. 5. The DFT-calculated potential energy profiles on the reactions of lower-lying CuC3H− isomers with CO. The relative energies are in unit of eV and the bond lengths are given in pm. The structures of intermediates and transition states are plotted.
Fig. 6. (A) Spin crossing from the triplet state to the singlet state. The curve for the triplet state is from the intrinsic reaction coordinate (IRC) calculations. The energies of the singlet states are calculated at the IRC determined triplet state structures. (B) Spin crossing from the quartet state to the doublet state. The curve for the quartet state is from the Scan calculation. The energies of the doublet states are calculated at the Scan determined quartet state structures.


Set citation alerts for the article
Please enter your email address

© Copyright 2018-2021 | Chinese Laser Press. All Rights Reserved 沪ICP备15018463号-20