 AI Video Guide
AI Video Guide  AI Picture Guide
AI Picture Guide AI One Sentence
AI One Sentence

Philip Dalladay-Simpson, Bartomeu Monserrat, Li Zhang, Federico Gorelli. Distinct vibrational signatures and complex phase behavior in metallic oxygen[J]. Matter and Radiation at Extremes, 2024, 9(2): 028401
- Matter and Radiation at Extremes
- Vol. 9, Issue 2, 028401 (2024)
Note: This section is automatically generated by AI . The website and platform operators shall not be liable for any commercial or legal consequences arising from your use of AI generated content on this website. Please be aware of this.
Abstract
I. INTRODUCTION
Pressure is a fundamental thermodynamic tool to probe and tune novel interactions within materials by driving a reduction in atomic and molecular volumes. This is perhaps most clearly demonstrated in molecular systems, where decreasing the distance between molecular units can enhance intermolecular interactions and, under sufficient compression, can render familiar ambient dielectric gases into metals1–3 and/or close packed atomic solids.4 A prominent example is metallic-atomic solid hydrogen, originally proposed by Wigner and Huntington in 1935,2 which is expected to exist at densities just beyond those achievable with current experimental techniques and has come to be known as a “holy grail” of condensed matter sciences. By contrast, oxygen, a fundamental molecule with high terrestrial abundance, metallizes at relatively routine pressures (about one million atmospheres) and has become the prototypical example of density-driven molecular metallization. Over 30 years ago, Desgreniers et al.1 reported the first evidence for the metallization of oxygen, observing a significant enhancement of reflectivity in the visible and near-infrared. They also noted at their highest pressures, about 130 GPa, that oxygen becomes visually indistinguishable from the surrounding metallic gasket material. This metallic behavior in oxygen was subsequently confirmed by resistivity measurements5 and, shortly afterward, superconductivity was observed at about 100 GPa and 0.5 K.5 More recently, the metallic character of oxygen has been confirmed by x-ray Raman scattering measurements at a comparable 94 GPa.6 The metallization of oxygen was reported to be accompanied by an isostructural transition with abrupt changes in x-ray diffraction,7 which is at odds with metallization in other diatomic molecular systems, such as the halogens and hydrogen halides, which typically undergo a second-order metallization with no structural modification, as is also expected to be the case for hydrogen.4,8–10
From a historical standpoint, a period of three decades elapsed from the discovery in 1979 of the lower-pressure ϵ-O2 phase to its full structural refinement, with the reported observation of O8 clusters in the form of (O2)4 units.11,12 As far as structural characterization of the metallic ζ-O2 phase is concerned, this timespan has now been surpassed, and therefore fundamental physical insight into the nature of metallic oxygen has remained elusive. Currently, our best understanding of this phase comes from the interplay of numerical simulations, specifically ab initio evolutionary crystal structure predictions,13 and experimental observations.7,14–16 Simulations have yielded two energetically competitive monoclinic structures with similar enthalpies and with space groups C2/c and C2/m with four and eight molecules per unit cell, respectively.13 Of these two, the C2/m structure, which is isostructural to the ϵ-O2 phase, was identified as the most promising candidate structure, since it better reproduced the experimentally observed powder x-ray diffraction (XRD) measurements7 and Raman frequencies.15 This C2/m structure retains the (O2)4 molecular units of the precursory ϵ-O2 phase, and therefore no significant molecular rearrangement is expected for the ϵ–ζ phase transition. Further support for this choice of space group came from subsequent single-crystal diffraction results for ζ-O2 at 133 GPa;14 although the authors of Ref. 14 could index both a C2/c and an a C2/m unit cell, the latter was chosen on the basis of a complex analysis of crystal twinning and the number of modes present in the recorded Raman spectrum.
Raman spectroscopy has long been a pioneering tool for mapping out vast swathes of phase space for materials, particularly for molecular systems. This is well exemplified by the complex low-pressure phase diagram of oxygen, where the molecule’s magnetic moment plays a significant role. At higher pressures, the precise nature of the phase diagram around the metallization of oxygen remains poorly constrained, in part because of the exceptionally challenging nature of measuring the Raman effect in metals,17 which is nontrivial even under ambient conditions.18 This difficulty is due to the presence of free charge carriers, which can effectively screen incident electromagnetic radiation, as a consequence of which depths of only tens of nanometers can be probed with conventional Raman spectroscopy. Owing to these challenges, Raman studies of metals at high pressures have remained limited compared with other classes of materials, with only a few experiments reported on the metallic oxygen system to date.14,15,19
In the study described in the present paper, oxygen was loaded into a diamond anvil cell and compressed to pressures in excess of two million atmospheres in order to characterize the Raman signature of ζ-O2 as well as the phase diagram of O2 at extreme densities. To rule out the possibility of interference by chemical reactions, various methods for loading oxygen samples were adopted, such as (1) O2 in helium, (2) O2 in helium with laser heating, and (3) compression of bulk O2 (more details of experimental loading procedures can be found in the
II. RESULTS AND DISCUSSION
A. Raman signature of
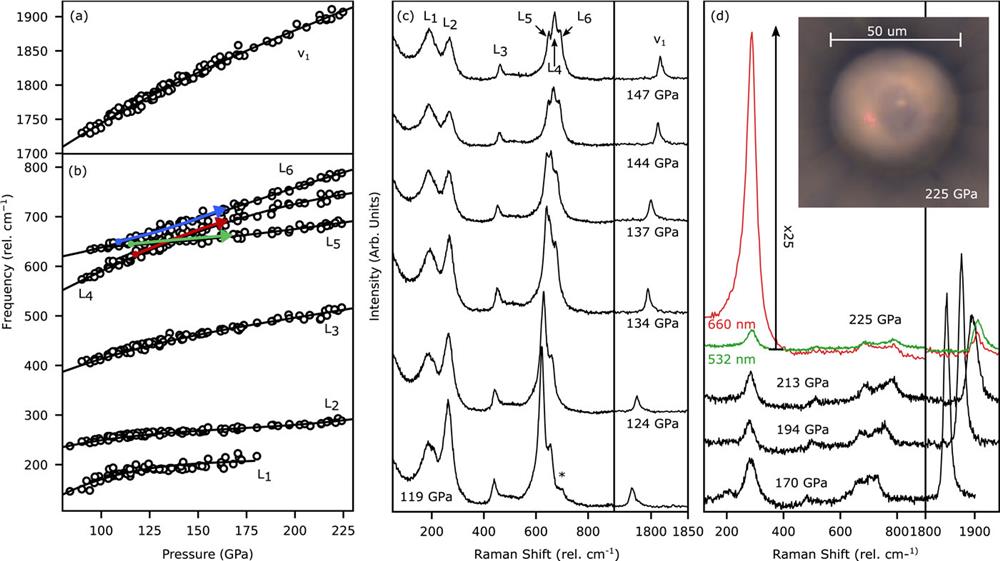
Here, ζ-O2 was observed from 85 GPa up to the highest pressures of 225 GPa, as can be seen in Figs. 1 and 5 and in Fig. S2 (
![]()
Figure 1.Raman spectra of
Perhaps the most striking property of the vibrational spectrum of ζ-O2 is its simplicity when compared with the precursory phase ϵ-O2. Here, only seven excitations, namely, six lattice modes (L1, …, L6) and one vibrational mode (ν1), can be attributed to ζ-O2. This is in stark contrast to the ϵ-O2 phase, which has as many as 12 Raman-active modes,13,15,19 which include an additional vibrational mode (ν2). Typically, the reduction in the number of modes is indicative of a transition to a more simple structure, either to a higher symmetry Bravais lattice or a reduction in the number of molecules in the unit cell. The oxygen vibrational mode ν1 of ζ-O2 was found to coexist and be downshifted by only 9 cm−1 (0.5%) relative to the corresponding mode in the precursory ϵ-O2 phase and has a remarkably similar pressure dependence, as can be seen in Fig. S3 (
As the distinct vibrational spectra and pressure evolution of ζ-O2, as seen in Fig. 1, have now been constrained, it becomes possible to compare with energetically competitive candidate models. This has been attempted before,13,14 but owing to the coexistence of precursory phases and the broad/weak Raman response of metallic oxygen, it is challenging to make robust comparisons. For reference, Fig. S2(c) (
![]()
Figure 2.
The previously proposed C2/m structure (isostructural to ϵ-O2) for ζ-O2, has oxygen atoms occupying 8
B. Evidence for the dissociation of (O
The ultimate fate of O8 has given rise to significant speculation. O8 clusters, (O2)4 units, are a beautiful feature in the condensed oxygen system,23 and despite not being anticipated by theory, they demonstrate remarkable stability. These (O2)4 units are found to be stable from 7.6 GPa at 0 K24 to temperatures in excess of 750 K from 44 to 90 GPa25,26 and up to at least 96 GPa at 300 K,7,16 making them perhaps the most prominent feature of the known oxygen phase diagram. The mechanism behind the formation of these clusters, believed to be of Peierls distortion in origin,23 induces a weak chemical bond between a quartet of neighboring O2 molecules forming a rhomboid.11,12 Their remarkable stability on compression is confirmed by a substantial volume reduction of 5.4% with respect to the precursory phase, δ-O2.27 However, on compression, intercluster distances have been reported to become comparable to intracluster distances found at lower pressures,11 which could have profound implications for the stability and suitability of these clusters at higher pressures and in denser O2 phases.
The preservation of (O2)4 units in the previously proposed structure for ζ-O2, C2/m, is a curious conjecture. In this structure, although it still has four molecules in the primitive unit cell and the same symmetry as ϵ-O2, the shortest intercluster distance, x1 = 2.009 Å, has become shorter than the longest intracluster distance, x2 = 2.03 Å, at 130 GPa,13 as seen in Fig. S4 (
Arguably, Raman spectroscopy played the most significant role in the previous assignment of the C2/m structure to the ζ-O2 phase,13–15 largely on the basis of the number of apparent Raman modes present. Here, it should be noted that this established interpretation is compromised owing to the strong coexistence with the precursory ϵ-O2 phase and the nature of the phase transition as described in detail in Sec. II C. However, although the now distinct spectrum of ζ-O2 is much simpler, with only seven modes up to 175 GPa, the C2/m structure is the only candidate structure that provides a satisfactory explanation of all the modes observed in our experiments. Above 175 GPa, the observed Raman spectrum becomes entirely compatible with the expected number of modes and energies from the calculated C2/c and Ci structures, owing to the loss of L1. Therefore, at this pressure, it is unlikely that any vestiges of the (O2)4 clusters that are characteristic of the ϵ-O2 phase and responsible for the C2/m assignment are still present, since these clusters will have become completely dissociated. In addition, at this pressure, the precursory phase, ϵ-O2, is no longer detectable. This interpretation is substantiated by calculations showing that the C2/m structure is distorted to Ci; this instability can be seen in the simulated Raman modes in Fig. 2(a), where above 125 GPa the lowest-frequency Raman mode of the C2/m structure is found to turn over and rapidly decrease in frequency, becoming zero at 210 GPa. Further, we can see from the calculations that this transition is very subtle and is particularly difficult to verify experimentally, with all phases demonstrating similar Bragg reflections and Raman activity, as seen in Fig. 2.
Perhaps related to this phase transition is an observed Raman resonance of the L2 mode with excitation energy 1.88 eV (660 nm), where an enhancement of up to 25 times at 225 GPa is reached compared with the Raman excitation by 2.33 eV (532 nm), as seen in Fig. 1(d). This observed resonance results in a substantial Raman response for a metallic system, since the L2 mode is found to be more intense than the unstressed diamond optical phonon and 84 times stronger than the optical phonon of the surrounding metallic (rhenium) gasket. Raman resonance, although unusual in metallic systems, is not uncommon in molecular systems in the vicinity of an insulator-to-metallic transition and has been observed in hydrogen in its semiconducting states9,28–30 en route to its elusive metallic form.2 The resonance found in metallic oxygen, which was found to increase with pressure above 150 GPa, is a clear indication that although the system remains metallic, there is a continued modification in the underlying electronic structure.
At pressures above 175 GPa, it is also seen from numerical calculations that the Raman-active phonons assigned to L2 are found to soften, and if this trend were to continue, then these phonons would become imaginary at higher pressures. This behavior, which was not observed experimentally, is indicative that yet another phase transition could be expected at higher pressures. Recent calculations and experimental observations suggest that the high temperature η′-O2 (P63/mmc) phase,25,26 found at lower pressures, could be stable at room temperature at these pressures. Further, in agreement with this interpretation, a P63/mmc O2 phase was reported to be energetically competitive at 0.5 TPa20 and would act as the precursor to polymeric oxygen states in which the O2 molecule is finally broken.
C. Complex phase behavior
In all experiments, the evolution of the oxygen Raman spectrum at room temperature up to 130 GPa is consistent with previous reports,14,15,19,23 as can be seen in Fig. S2 (
![]()
Figure 3.Raman map of an originally
Above 100 GPa, new weak and broad Raman bands emerge and are found to coexist with modes attributed to ϵ′-O2. These features, which have been reported previously, have been assigned to ζ-O2, implying that the process of metallization is not associated with molecular dissociation.15 Unfortunately, owing to the strong overlap with the coexisting ϵ′-O2 phase and the weak Raman response from metals, these modes have remained poorly constrained. At pressures above 110 GPa, remarkable changes are found when metallic oxygen undergoes laser heating. These transformations, which are shown in Fig. 4, are observed both visually and spectroscopically. In the process of laser heating, a clear change was observed in the sample morphology, which turned from well-defined oxygen crystallites into a fine powder dispersed within the surrounding helium pressure medium, as shown in Fig. 4. This change in morphology occurred with no visible black body emission and relatively low laser powers (10 W), suggesting that annealing temperatures remained below 1000 K and therefore did not reach the melting conditions for either oxygen or the surrounding helium. After relatively moderate laser heating, a significant enhancement of the Raman signal was observed, with the formerly weak and broad excitations attributed to ζ-O2 becoming sharp and well resolved, as can be seen in Fig. 4(a), while overlapping peaks assigned to ϵ′-O2 were lost. Thus, the distinct vibrational signatures of ζ-O2, shown in Figs. 1 and 2, and of ζ-O2 could be seen. The enhancement of the Raman response can be correlated with the change in sample morphology, for two main reasons: (1) the suspension of a fine powder in helium will remove local deviatoric stresses on the oxygen crystals, which may broaden/weaken Raman excitations, and (2) the surface area, to which the Raman probe is sensitive, increases substantially. A similar Raman enhancement correlated with diminishing particle sizes has previously been reported in crystalline powders and has been largely attributed to a diffuse scattering mechanism.32,33 The profound change in sample morphology is intriguing and indicative of significant rearrangements of the oxygen molecules and therefore the associated crystalline domains. This unusual behavior is likely a consequence of the rapid dissociation and recombination of (O2)4 units from the excursion to the high-temperature η′-O2 phase during laser heating, as has been reported previously at lower pressures.25 The novelty and unprecedented quality of the ζ-O2 Raman spectrum can provide profound new insights into the elusive nature of metallic oxygen and has been discussed in Sec. II B.
![]()
Figure 4.Effects of laser heating on coexistence of
Compression along the room temperature isotherm to pressures above 175 GPa also results in a Raman spectrum of ζ-O2, with no modes characteristic of ϵ′-O2 being observed. In addition, the Raman responses from these experiments on decompression are identical to those from the previously discussed laser-heated samples, as seen in Fig. 3 and in Fig. S2 (
The observed strong coexistence between the ϵ′-O2 and ζ-O2, which almost doubles the known stability field of the precursory ϵ(ϵ′)-O2 phase, and the behavior on laser heating, can give insight into the nature of the ϵ′–ζ transition. This complex phase behavior, summarized in Fig. 5, is indicative of a first-order/structural transition, which is at odds with the concepts of displacive and/or second-order transitions that were previously used to describe the nature of the ϵ′–ζ transition.14,16 However, whether the ϵ′–ζ phase transition is isostructural, involving the C2/m phase as suggested by calculations,13 and the fate of the fascinating (O2)4 units, remain to be established, as discussed previously.
![]()
Figure 5.Hysteresis and coexistence of
III. CONCLUSION AND OUTLOOK
We have conducted a systematic study of oxygen up to 225 GPa through (1) compression of O2 in helium, (2) compression of O2 in helium with laser heating, and (3) compression of bulk O2. Through the course of these experiments, we have identified a strong metastability with precursory ϵ′-O2, which appears to be preserved up to pressures of 175 GPa and, critically, could have affected the interpretation of existing diffraction data.14,16 For the first time since the discovery of ζ-O2 over 30 years ago, we have recorded its distinct vibrational signature, which can be found either through the compression of oxygen to pressures greater than 175 GPa at room temperature or via laser heating oxygen crystals in helium at pressures greater than 110 GPa. Through a comparison with our numerical simulations, the obtained vibrational frequencies for ζ-O2 agree well with the C2/m candidate structure. However, at higher pressures, above 175 GPa, simpler candidate structures with only two molecules in their primitive unit cells, C2/c and Ci, appear to be more appropriate, enticingly suggesting a density-driven dissociation of the (O2)4 units. Finally, the mapped phase behavior and strong coexistence found in the metallic phases will help guide future studies of the physical properties of these phases and perhaps the long-awaited structural refinement of ζ-O2.
SUPPLEMENTARY MATERIAL
ACKNOWLEDGMENTS
Acknowledgment. We would like to thank Huichao Zhang, Huixin Hu, and Yuchen Yu for their assistance in the laboratory and during sample loadings, as well as Eugene Gregoryanz for useful discussions. Bartomeu Monserrat acknowledges support from a UKRI Future Leaders Fellowship (Grant No. MR/V023926/1), from the Gianna Angelopoulos Programme for Science, Technology, and Innovation, and from the Winton Programme for the Physics of Sustainability. Federico Gorelli acknowledges support from the Shanghai Science and Technology Committee, China (Grant No. 22JC1410300) and the Shanghai Key Laboratory of Novel Extreme Condition Materials, China (Grant No. 22dz2260800). Portions of this work were performed on beamline ID27 at the European Synchrotron Radiation Facility (ESRF) (Proposal Nos. CH6636 and CH6300), Grenoble, France as well as preliminary investigations on beamline 15U1 at the Shanghai Synchrotron Radiation Facility (SSRF), Shanghai, People’s Republic of China. We would like to thank our beamline contacts Mohamed Mezouar and Gaston Garbarino for their assistance during the course of the x-ray diffraction data collection. The computational resources were provided by the Cambridge Tier-2 system operated by the University of Cambridge Research Computing Service and funded by the UK EPSRC (Grant No. EP/P020259/1).
References
[1] S.Desgreniers, A. L.Ruoff, Y. K.Vohra. J. Phys. Chem., 94, 1117(1990).
[2] H. B.Huntington, E.Wigner. J. Chem. Phys., 3, 764(1935).
[3] A. S.Balchan, H. G.Drickamer. J. Chem. Phys., 34, 1948(1961).
[4] F.Hiroshi, T.Kenichi, S.Kyoko, O.Mitsuko. Nature, 423, 971(2003).
[5] K.Amaya, M. I.Eremets, M.Ikumo, K.Shimizu, K.Suhara. Nature, 393, 767(1998).
[7] Y.Akahama, M.Hanfland, D.H?usermann, H.Kawamura, O.Shimomura. Phys. Rev. Lett., 74, 4690(1995).
[9] A. P.Drozdov, M. I.Eremets, P. P.Kong, H.Wang. Nat. Phys., 15, 1246(2019).
[12] S.Desgreniers, P.Loubeyre, L. F.Lundegaard, M. I.McMahon, G.Weck. Nature, 443, 201(2006).
[13] C. W.Glass, Y.Ma, A. R.Oganov. Phys. Rev. B, 76, 064101(2007).
[14] S.Desgreniers, P.Loubeyre, M.Mezouar, G.Weck. Phys. Rev. Lett., 102, 255503(2009).
[16] R.LeToullec, P.Loubeyre, G.Weck. Phys. Rev. Lett, 88, 035504(2002).
[17] E.Burstein, A. A.Maradudin, D. L.Mills. Ann. Phys., 56, 504(1970).
[18] M.Ashkin, D. W.Feldman, J. H.Parker. Phys. Rev. Lett., 21, 607(1968).
[19] Y.Akahama, H.Kawamura. Phys. Rev. B, 54, R15602(1996).
[22] W. A.Bassett, S. H.Shim, C. S.Zha. Rev. Sci. Instrum., 75, 2409(2004).
[23] Y.Crespo, Y. A.Freiman, H. J.Jodl. Phys. Rep., 743, 1(2018).
[24] W. B.Carter, D.Gonzales, M. L.Lowe, D.Schiferl. J. Phys. Chem., 95, 2516(1991).
[26] S. A.Bonev, S. F.Elatresh. Phys. Chem. Chem. Phys., 22, 12577(2020).
[27] R. L.Mills, B.Olinger, R. B.Roof. J. Chem. Phys., 81, 5068(1984).
[28] P.Dalladay-Simpson, E.Gregoryanz, R. T.Howie. Nature, 529, 63(2016).
[31] S.Desgreniers, P.Loubeyre, G.Weck.
[32] C. K.Mann, T. J.Vickers, H.Wang. Appl. Spectrosc., 56, 1538(2002).
[33] P. J.Hendra, R. J.Lehnert, M. V.Pellow-Jarman. Vib. Spectrosc., 12, 257(1996).


Set citation alerts for the article
Please enter your email address

© Copyright 2018-2021 | Chinese Laser Press. All Rights Reserved 沪ICP备15018463号-20