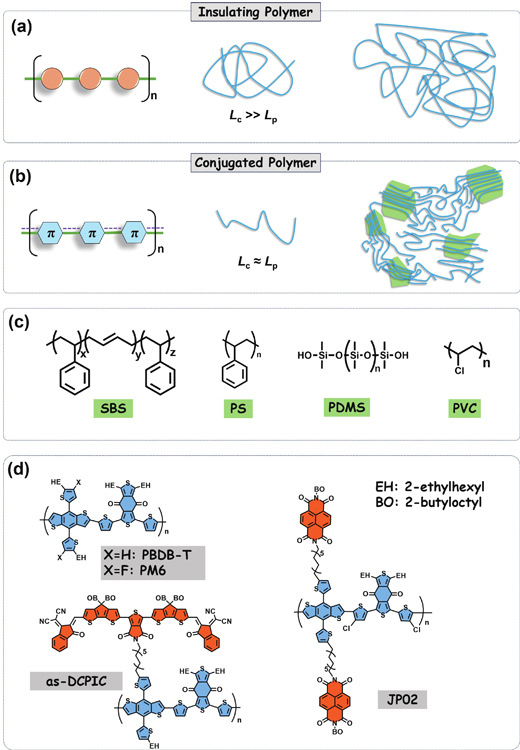
Figure 1.(Color online) The bonding, molecular conformation and film morphology for the insulating polymers (a) and conjugated polymers (b). (c) Chemical structures for SBS, PS, PDMS, and PVC. (d) Chemical structures for PBDB-T, PM6, as-DCPIC, and JP02.
Recently, Li et al. uncovered the “non-covalent bonding” between polyvinyl chloride (PVC) and PM6[10]. This interaction enhances the molecular-level compatibility in this system and addresses the miscibility issue. The interaction was precisely identified through the tactic segment of PVC and the carbonyl units. PM6/PVC films exhibit an improved crack-onset strain of 19.35%, as compared to 10.12% for pristine PM6 film. The generality of this strategy was validated in various polymers, such as PBDB-T and as-DCPIC[11] (Fig. 1(d)) with carbonyl units. These works will help to select molecular interactions to avoid the miscibility issue, thus improving the photovoltaic and mechanical stability. This study will favor the application of conjugated polymers in flexible organic electronics.
Wearable electronics require the stretchability of conjugated polymers. This has prompted the exploration of combining insulating and conjugated polymers to achieve both high charge transport and mechanical resilience. A variety of insulating polymers (Fig. 1(c)), such as polydimethylsiloxane (PDMS), polystyrene-block-polybutylene-block-polystyrene block copolymer (SBS), and polystyrene (PS) with high flexibility, have been applied into insulating/conjugated polymer blends. However, they face the limited compatibility with all conjugated polymers. For instance, PS may be suitable for double-cable conjugated polymer JP02[8] (Fig. 1(d)) but unsuitable for PM6[9], so the universality becomes an issue.
Insulating polymers are characterized by a predominantly σ-covalent structure, which localize electrons in the atoms and exhibit dielectricity. Insulating polymers typically adopt a more linear and extended conformation, as the repeating units are connected by single covalent bonds, resulting in a relatively straight and extended chain structure. For most insulating polymers, the contour length (Lc) is significantly larger than their persistence length (Lp) due to the rotation of C−C single bonds (Fig. 1(a)). Consequently, this leads to a flexible, random-coil chain conformation. This structural feature contributes to the great mechanical durability and resistance to crack initiation during stretching or bending processes. In contrast, conjugated polymers possess a π-conjugated molecular structure, allowing electron mobility along the main chain, called delocalization, which imparts semiconducting properties[1, 2]. The presence of rigid, alternating single and multiple bonds results in comparable Lc and Lp, thereby yielding a stiff or semi-flexible conformation (Fig. 1(b))[3, 4]. As a consequence, most conjugated polymers are prone to fracture under low strain levels (<10%)[5−7].