
Qianrui Liu, Junyi Li, Mohan Chen. Thermal transport by electrons and ions in warm dense aluminum: A combined density functional theory and deep potential study[J]. Matter and Radiation at Extremes, 2021, 6(2): 026902
- Matter and Radiation at Extremes
- Vol. 6, Issue 2, 026902 (2021)
Abstract
I. INTRODUCTION
Warm dense matter (WDM) is a state of matter lying between condensed matter and plasma and consisting of strongly coupled ions and partially degenerate electrons. WDM exists in the interiors of giant planets
The thermal conductivity includes both electronic and ionic contributions. The Kubo–Greenwood (KG) formula
While current experimental techniques only measure the total thermal conductivity, first-principles methods have become ideal tools to yield electronic and ionic contributions separately. However, only a few studies adopting first-principles methods have examined transport properties by considering both contributions.
In this work, we demonstrate that the DP method in conjunction with DFT can be used to obtain both electronic and ionic thermal conductivities of warm dense Al. We use KSDFT, OFDFT, and DPMD to study the electronic and ionic thermal conductivities of warm dense Al at temperatures of 0.5 eV, 1.0 eV, and 5.0 eV with a density of 2.7 g/cm3. The electronic thermal conductivity can be accurately computed via the KG method based on the DPMD trajectories instead of the FPMD trajectories. Importantly, we systematically investigate convergence issues with regard to the number of k-points, the number of atoms, the broadening parameter, the exchange-correlation functionals, and the pseudopotentials, together with their effects on the determination of the electronic thermal conductivity via DPMD simulations. Furthermore, the ionic thermal conductivity can also be obtained via DPMD simulations, and we study convergence for different sizes of systems and different lengths of trajectories.
The remainder of the paper is organized as follows. In Sec.
II. METHOD
A. Density functional theory
The ground-state total energy within the formalism of DFT
We run 64-atom Born–Oppenheimer molecular dynamics (BOMD) simulations with KSDFT using the
We also perform 108-atom BOMD simulations with OFDFT, using the PROFESS 3.0 package.
B. Deep potential molecular dynamics
The DP method
In this work, we adopt DNN-based models trained from either KSDFT or OFDFT trajectories with the DeePMD-kit package.
C. Kubo–Greenwood formula
The electronic thermal conductivity κe is calculated from the Onsager coefficients Lmn as
The KG method needs eigenvalues and wave functions computed from DFT solutions of given atomic configurations. In practice, we select 5–20 atomic configurations from the last 2 ps MD trajectories, with a time interval of 0.1 ps. We use both PBE and LDA XC functionals and the associated norm-conserving (NC) pseudopotentials to examine the influences of XC functionals and pseudopotentials on the resulting electronic thermal conductivity. We adopt two NC pseudopotentials for Al, which are referred to here as PP1 and PP2. The PP1 pseudopotential is generated with the optimized norm-conserving Vanderbilt pseudopotential method via the ONCVPSP package.
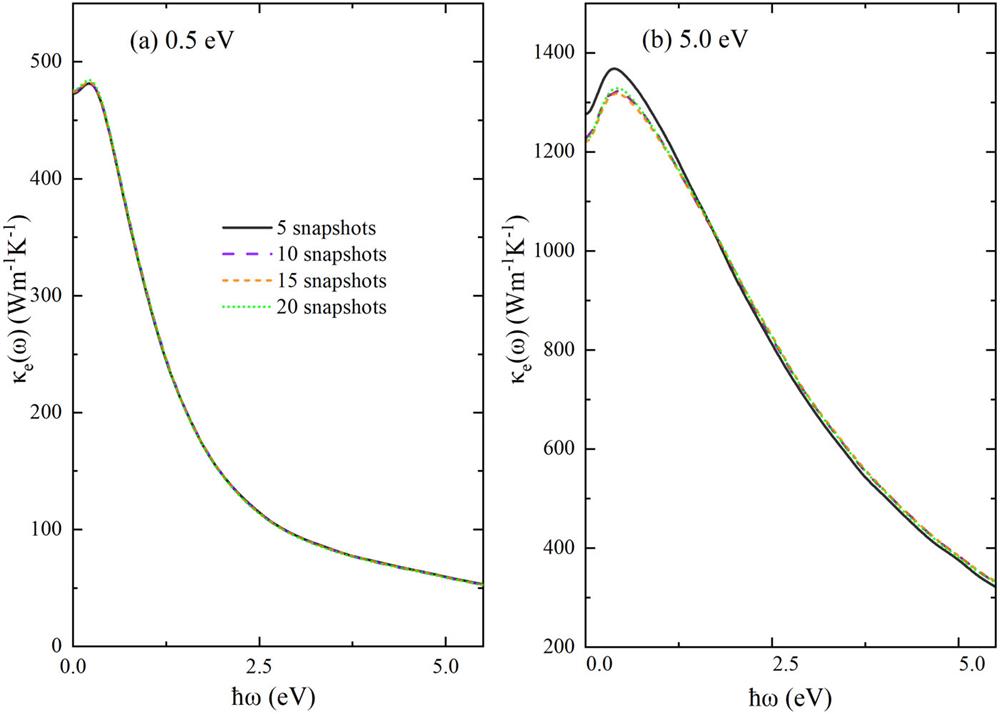
![]()
Figure 1.Convergence of frequency-dependent electronic thermal conductivity
D. Green–Kubo formula
In the DPMD method, the total potential energy of the system is decomposed onto each atom. In this regard, the ionic thermal conductivity can be calculated through the GK method
Equation
III. RESULTS
A. Accuracy of DP models
We first perform FPMD simulations of Al based on KSDFT and OFDFT at temperatures of 0.5 eV, 1.0 eV, and 5.0 eV. The PBE XC functional is used, and the FPMD trajectory length is 10 ps. The cell contains 64 Al atoms. Two DP models, named DP-KS and DP-OF, are trained, based on the KSDFT and OFDFT trajectories, respectively. Note that the accuracy of the DP models in describing warm dense Al at the three temperatures has been demonstrated in our previous work,
Here, we first focus on the frequency-dependent Onsager coefficients Lmn(ω).
![]()
Figure 2.Frequency-dependent Onsager kinetic coefficients (a)
The results for the frequency-dependent electronic thermal conductivity κe(ω) computed using the four different methods are illustrated in
![]()
Figure 3.Frequency-dependent electronic thermal conductivity
B. Convergence of electronic thermal conductivity
Previous work
1. Number of k-points
We first investigate the convergence of κe(ω) with respect to different numbers of k-points.
![]()
Figure 4.Convergence of frequency-dependent electronic thermal conductivity
| N | 0.5 eV | 1.0 eV | 5.0 eV |
|---|---|---|---|
| 16 | 8 × 8 × 8 | 7 × 7 × 7 | 3 × 3 × 3 |
| 32 | 6 × 6 × 6 | 5 × 5 × 5 | 3 × 3 × 3 |
| 64 | 3 × 3 × 3 | 3 × 3 × 3 | 1 × 1 × 1 |
| 108 | 2 × 2 × 2 | 2 × 2 × 2 | 1 × 1 × 1 |
| 216 | 2 × 2 × 2 | 2 × 2 × 2 | 1 × 1 × 1 |
| 256 | 2 × 2 × 2 | 2 × 2 × 2 | 1 × 1 × 1 |
| 432 | 2 × 2 × 2 | N/A | N/A |
Table 1. Sizes of k-points adopted in KSDFT calculations to converge the electronic thermal conductivity of Al with different numbers N of atoms in the simulation cell at temperatures of 0.5 eV, 1.0 eV, and 5.0 eV.
2. Number of atoms
The value of κe(ω) converges not only for a sufficient number of k-points but also for a sufficient number of atoms in the simulation cell. To demonstrate this, we plot κe(ω) with respect to different numbers of atoms in
![]()
Figure 5.Convergence of the electronic thermal conductivity
We perform a further analysis to elucidate the origin of the size effects in computations of κe(ω). As Eq.
To clarify this issue, we define an energy interval distribution function (EIDF) as
![]()
Figure 6.Density of states of a 256-atom cell at temperatures of (a) 0.5 eV and (b) 5.0 eV. The Fermi–Dirac function at the same temperature is plotted as a black solid line. The DP-OF model refers to the DP model trained from OFDFT molecular dynamics trajectories.
![]()
Figure 7.Energy interval distribution function of different cells at (a) 0.5 eV and (b) 5.0 eV. Bands within 6.0 eV and 50.5 eV above the chemical potential
3. Broadening parameter
The FWHM broadening parameter σ that appears in the δ(E) function in Eq.
![]()
Figure 8.(a) Energy interval distribution function and (b) electronic thermal conductivity of a 256-atom cell at 0.5 eV. The snapshots are from DPMD simulations. The DPMD model is trained from OFDFT trajectories with the PBE XC functional. Different line styles represent different values of the broadening parameter
![]()
Figure 9.Electronic thermal conductivities
4. Exchange-correlation functionals
We study the influences of the LDA and PBE XC functionals on the computed κe(ω) by first validating the atomic configurations generated by FPMD simulations. Specifically, atomic configurations are chosen from two 256-atom DPMD trajectories, which are generated by two DP models trained from OFDFT with the LDA and PBE XC functionals. We then adopt the KG method using the PP1 pseudopotentials generated with the same XC functional and obtain κe at temperatures of 0.5 eV, 1.0 eV, and 5.0 eV. As shown in
| T (eV) | κe(PP1) | κe(PP2) | κI | |
|---|---|---|---|---|
| 0.5 | 485.1 | 486.6 | 1.422 ± 0.034 | |
| DP-OF (PBE) | 1.0 | 764.1 | 772.3 | 1.469 ± 0.086 |
| 5.0 | 1281.7 | 1604.6 | 2.091 ± 0.031 | |
| 0.5 | 477.3 | 475.6 | 1.394 ± 0.047 | |
| DP-OF (LDA) | 1.0 | 773.8 | 779.0 | 1.318 ± 0.032 |
| 5.0 | 1246.4 | 1568.5 | 2.141 ± 0.051 | |
| 0.5 | 466.5 | 1.419 ± 0.038 | ||
| DP-KS (PBE) | 1.0 | 771.0 | 1.393 ± 0.066 | |
| 5.0 | 1305.8 | 2.075 ± 0.066 |
Table 2. Electronic thermal conductivity κe and ionic thermal conductivity κI (both in units of W m−1 K−1) at temperatures T of 0.5 eV, 1.0 eV, and 5.0 eV. The results are computed from the DP-KS and DP-OF molecular dynamics trajectories. DP-KS and DP-OF refer to the DP models trained from KSDFT and OFDFT molecular dynamics trajectories, respectively.
5. Pseudopotentials
We investigate how norm-conserving pseudopotentials affect the computed electronic thermal conductivity κe. First of all,
We find the following. First, the DP-OF results agree reasonably well with the DP-KS ones, as has previously been shown in
C. Ionic thermal conductivity
The ionic thermal conductivity of warm dense Al can be evaluated using the GK formula, since the atomic energies are available in the DPMD method. However, the computed ionic thermal conductivity may be affected by trajectory length and system size. We therefore study the convergence of the ionic thermal conductivity with respect to different lengths of trajectories and system sizes. We first test the convergence of the autocorrelation function CJ(t) in Eq.
![]()
Figure 10.Autocorrelation function of heat current
Next, to examine size effects on the ionic thermal conductivity, we run simulations for 500 ps for 12 different system sizes, with the number of atoms per cell ranging from 16 to 65 536. The results are shown in
![]()
Figure 11.Computed ionic thermal conductivity of warm dense Al at (a) 0.5 eV, (b) 1.0 eV, and (c) 5.0 eV for 12 different system sizes. The numbers of atoms per cell in these systems are
IV. CONCLUSIONS
We have proposed a method that combines DPMD and DFT to calculate both electronic and ionic thermal conductivities of materials, with the DP models being trained from DFT-based MD trajectories. The resulting DP models accurately reproduce the properties predicted by DFT. In addition, they can be utilized to efficiently simulate a large cell consisting of hundreds of atoms, which largely mitigates the size effects caused by periodic boundary conditions. By taking the atomic configurations from DPMD trajectories, we can use the eigenvalues and eigenstates of a given system obtained from DFT solutions and employ the KG formula to compute the electronic thermal conductivity. In addition, the DP models yield atomic energies, which are not available in the traditional DFT method. By using the atomic energies to evaluate the ionic thermal conductivity, both electronic and ionic contributions to the thermal conductivity can be obtained for a given material.
We took warm dense Al as an example and thoroughly studied its thermal conductivity. Expensive FPMD simulations of large systems can be replaced by DPMD simulations with much smaller computational resources. We first computed the temperature-dependent electronic thermal conductivities of warm dense Al from 0.5 eV to 5.0 eV at a density of 2.7 g/cm3 with snapshots from OFDFT, KSDFT, and DPMD, and the three methods yielded almost the same results, demonstrating that the DPMD method has similar accuracy to FPMD simulations. We then systematically investigated convergence issues with respect to the number of k-points, the number of atoms, the broadening parameter, the exchange-correlation functionals, and the pseudopotentials. A 256-atom system was found to be large enough to converge the electronic thermal conductivity. The broadening parameter was chosen to be 0.4 eV according to our analysis of the energy interval distribution function. We found that both LDA and PBE XC functionals yielded similar results for the electronic thermal conductivity. However, the choice of pseudopotentials may substantially affect the resulting electronic thermal conductivity. We also computed the ionic thermal conductivity with DPMD and the GK method and investigated convergence issues with respect to trajectory length and system size. We found that the ionic thermal conductivity of warm dense Al was much smaller than its electronic thermal conductivity. In summary, the DPMD method provides promising accuracy and efficiency in studying both electronic and ionic thermal conductivities of warm dense Al and should be considered for future work on modeling transport properties of WDM.
References
[1] T. Guillot. Interiors of giant planets inside and outside the solar system. Science, 286, 72-77(1999).
[2] B. Holst, A. Becker, N. Nettelmann et al. Jupiter models with improved ab initio hydrogen equation of state (H-REOS.2). Astrophys. J., 750, 52(2012).
[3] F. Wesemael, M. P. Savedoff, H. M. Van Horn et al. Atmospheres for hot, high-gravity stars. I-Pure hydrogen models. Astrophys. J., Suppl. Ser., 43, 159(1980).
[4] J. Daligault, S. Gupta. Electron-ion scattering in dense multi-component plasmas: Application to the outer crust of an accreting neutron star. Astrophys. J., 703, 994-1011(2009).
[5] R. LeToullec, D. Hausermann, P. Loubeyre et al. X-ray diffraction and equation of state of hydrogen at megabar pressures. Nature, 383, 702-704(1996).
[6] W. J. Nellis, S. T. Weir, A. C. Mitchell. Metallization of fluid molecular hydrogen at 140 GPa (1.4 Mbar). Phys. Rev. Lett., 76, 1860-1863(1996).
[7] W. J. Nellis. Dynamic compression of materials: Metallization of fluid hydrogen at high pressures. Rep. Prog. Phys., 69, 1479-1580(2006).
[8] R. Cauble, L. B. Da Silva, T. S. Perry et al. Absolute measurements of the equations of state of low-Z materials in the multi-Mbar regime using laser-driven shocks. Phys. Plasmas, 4, 1857-1861(1997).
[9] J. Lindl. Development of the indirect-drive approach to inertial confinement fusion and the target physics basis for ignition and gain. Phys. Plasmas, 2, 3933-4024(1995).
[10] W. Kohn, P. Hohenberg. Inhomogeneous electron gas. Phys. Rev., 136, B864(1964).
[11] W. Kohn, L. J. Sham. Self-consistent equations including exchange and correlation effects. Phys. Rev., 140, A1133(1965).
[12] Y. A. Wang, E. A. Carter. Orbital-free kinetic-energy density functional theory. Theoretical Methods in Condensed Phase Chemistry, 117-184(2002).
[13] D. M. Ceperley, E. L. Pollock. Simulation of quantum many-body systems by path-integral methods. Phys. Rev. B, 30, 2555-2568(1984).
[14] D. M. Ceperley, E. L. Pollock. Path-integral computation of the low-temperature properties of liquid 4He. Phys. Rev. Lett., 56, 351-354(1986).
[15] E. W. Brown, J. L. DuBois, B. K. Clark et al. Path-integral Monte Carlo simulation of the warm dense homogeneous electron gas. Phys. Rev. Lett., 110, 146405(2013).
[16] K. P. Driver, B. Militzer. Development of path integral Monte Carlo simulations with localized nodal surfaces for second-row elements. Phys. Rev. Lett., 115, 176403(2015).
[17] R. Redmer, B. Holst, M. P. Desjarlais. Thermophysical properties of warm dense hydrogen using quantum molecular dynamics simulations. Phys. Rev. B, 77, 184201(2008).
[18] J.-P. Crocombette, V. Recoules.
[19] P. Zhang, C. Wang. Wide range equation of state for fluid hydrogen from density functional theory. Phys. Plasmas, 20, 092703(2013).
[20] T. Dornheim, M. Bonitz, Z. A. Moldabekov et al.
[21] W. B. Hubbard. Thermal structure of Jupiter. Astrophys. J., 152, 745-754(1968).
[22] S. C. Solomon, R. W. Siegfried. Mercury: Internal structure and thermal evolution. Icarus, 23, 192-205(1974).
[23] S. Labrosse. Thermal and magnetic evolution of the Earth’s core. Phys. Earth Planet. Inter., 140, 127-143(2003).
[24] M. M. Marinak, T. R. Dittrich, S. W. Haan et al. A comparison of three-dimensional multimode hydrodynamic instability growth on various National Ignition Facility capsule designs with HYDRA simulations. Phys. Plasmas, 5, 1125-1132(1998).
[25] L. V. Zhigilei, D. S. Ivanov. Combined atomistic-continuum modeling of short-pulse laser melting and disintegration of metal films. Phys. Rev. B, 68, 064114(2003).
[26] T. R. Boehly, S. X. Hu, L. A. Collins et al. First-principles thermal conductivity of warm-dense deuterium plasmas for inertial confinement fusion applications. Phys. Rev. E, 89, 043105(2014).
[27] R. Kubo. Statistical-mechanical theory of irreversible processes. I. General theory and simple applications to magnetic and conduction problems. J. Phys. Soc. Jpn., 12, 570-586(1957).
[28] D. A. Greenwood. The Boltzmann equation in the theory of electrical conduction in metals. Proc. Phys. Soc., 71, 585-596(1958).
[29] M. P. Desjarlais, L. A. Collins, J. D. Kress. Electrical conductivity for warm, dense aluminum plasmas and liquids. Phys. Rev. E, 66, 025401(2002).
[30] P. R Levashov, D. V. Knyazev.
[31] P. R. Levashov, D. V. Knyazev. Transport and optical properties of warm dense aluminum in the two-temperature regime:
[32] N. Nettelmann, J. Wicht, M. French, A. Becker, R. Redmer, W. Lorenzen, M. Bethkenhagen.
[33] V. Recoules, A. Decoster, F. Lambert et al. On the transport coefficients of hydrogen in the inertial confinement fusion regime. Phys. Plasmas, 18, 056306(2011).
[34] N. de Koker, G. Steinle-Neumann, V. Vlček. Electrical and thermal conductivity of Al liquid at high pressures and temperatures from ab initio computations. Phys. Rev. B, 85, 184201(2012).
[35] V. Recoules, C. E. Starrett, J. Clérouin et al. Average atom transport properties for pure and mixed species in the hot and warm dense matter regimes. Phys. Plasmas, 19, 102709(2012).
[36] S. X. Hu, V. N. Goncharov, B. Militzer et al. Strong coupling and degeneracy effects in inertial confinement fusion implosions. Phys. Rev. Lett., 104, 235003(2010).
[37] S. Crockett, D. Sheppard, J. D. Kress et al. Combining Kohn-Sham and orbital-free density-functional theory for Hugoniot calculations to extreme pressures. Phys. Rev. E, 90, 063314(2014).
[38] M. Pozzo, D. Alfè, M. P. Desjarlais. Electrical and thermal conductivity of liquid sodium from first-principles calculations. Phys. Rev. B, 84, 054203(2011).
[39] M. S. Green. Markoff random processes and the statistical mechanics of time-dependent phenomena. II. Irreversible processes in fluids. J. Chem. Phys., 22, 398-413(1954).
[40] D. A. McQuarrie. Statistical Mechanics, 520-521(1976).
[41] T. Kawamura, Y. Kangawa, K. Kakimoto. Investigation of thermal conductivity of gan by molecular dynamics. J. Cryst. Growth, 284, 197-202(2005).
[42] H. Rezania, F. Taherkhani. Temperature and size dependency of thermal conductivity of aluminum nanocluster. J. Nanopart. Res., 14, 1222(2012).
[43] W. K. Kim, J. H. Shim, M. Kaviany. Thermophysical properties of liquid UO2, ZrO2 and corium by molecular dynamics and predictive models. J. Nucl. Mater., 491, 126-137(2017).
[44] R. Ramprasad, C. Carbogno, M. Scheffler.
[45] P. Umari, S. Baroni, A. Marcolongo. Microscopic theory and quantum simulation of atomic heat transport. Nat. Phys., 12, 80-84(2016).
[46] J. Kang, L.-W. Wang. First-principles Green-Kubo method for thermal conductivity calculations. Phys. Rev. B, 96, 020302(2017).
[47] M. French. Thermal conductivity of dissociating water—An ab initio study. New J. Phys., 21, 023007(2019).
[48] A. Jain, A. J. H. McGaughey. Thermal transport by phonons and electrons in aluminum, silver, and gold from first principles. Phys. Rev. B, 93, 081206(2016).
[49] J. Ma, W. Li, Y. Chen. Understanding the thermal conductivity and Lorenz number in tungsten from first principles. Phys. Rev. B, 99, 020305(2019).
[50] H. Wang, L. Zhang, J. Han et al. End-to-end symmetry preserving inter-atomic potential energy model for finite and extended systems. Advances in Neural Information Processing Systems, 4436-4446(2018).
[51] J. Han, H. Wang, L. Zhang et al. DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics. Comput. Phys. Commun., 228, 178-184(2018).
[52] J. Han, H. Wang, L. Zhang et al. Deep potential molecular dynamics: A scalable model with the accuracy of quantum mechanics. Phys. Rev. Lett., 120, 143001(2018).
[53] R. Car, L. Lin, D. Lu, H. Wang, W. E, L. Zhang, W. Jia, M. Chen. Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning(2020).
[54] H. Wang, W. Jia, W. E, L. Lin, M. Chen, L. Zhang, R. Car, D. Lu. 86 PFLOPS deep potential molecular dynamics simulation of 100 million atoms with ab initio accuracy. Comput. Phys. Commun., 259, 107624(2021).
[55] M. Parrinello, L. Bonati. Silicon liquid structure and crystal nucleation from
[56] B. Wen, Y. Sun, F.-Z. Dai et al. Theoretical prediction on thermal and mechanical properties of high entropy (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C by deep learning potential. J. Mater. Sci. Technol., 43, 168-174(2020).
[57] L. Zhang, H.-Y. Ko, B. Santra et al. Isotope effects in liquid water via deep potential molecular dynamics. Mol. Phys., 117, 3269-3281(2019).
[58] J. Xu, L. Zhang, M. Chen, X. Wu, B. Santra, C. Zhang. Isotope effects in molecular structures and electronic properties of liquid water via deep potential molecular dynamics based on the scan functional. Phys. Rev. B, 102, 214113(2020).
[59] Q. Liu, M. Chen, D. Lu. Structure and dynamics of warm dense aluminum: A molecular dynamics study with density functional theory and deep potential. J. Phys.: Condens. Matter, 32, 144002(2020).
[60] C. Gao, Q. Liu, Y. Zhang et al. Warm dense matter simulation via electron temperature dependent deep potential molecular dynamics. Phys. Plasmas, 27, 122704(2020).
[61] L. H. Thomas. The calculation of atomic fields. Math. Proc. Cambridge Philos. Soc., 23, 542-548(1927).
[62] E. Fermi. Un metodo statistico per la determinazione di alcune priorieta dell’atome. Rend. Accad. Naz. Lincei, 6, 32(1927).
[63] E. Fermi. Eine statistische methode zur bestimmung einiger eigenschaften des atoms und ihre anwendung auf die theorie des periodischen systems der elemente. Z. Phys., 48, 73-79(1928).
[64] C. v. Weizsäcker. Zur theorie der kernmassen. Z. Phys. A: Hadrons Nucl., 96, 431-458(1935).
[65] M. P. Teter, L.-W. Wang. Kinetic-energy functional of the electron density. Phys. Rev. B, 45, 013196(1992).
[66] N. D. Mermin. Thermal properties of the inhomogeneous electron gas. Phys. Rev., 137, A1441(1965).
[67] T. Brumme, O. Andreussi, P. Giannozzi et al. Advanced capabilities for materials modelling with quantum ESPRESSO. J. Phys.: Condens. Matter, 29, 465901(2017).
[68] M. Ernzerhof, K. Burke, J. P. Perdew. Generalized gradient approximation made simple. Phys. Rev. Lett., 77, 3865(1996).
[69] P. E. Blöchl. Projector augmented-wave method. Phys. Rev. B, 50, 017953(1994).
[70] N. A. W. Holzwarth, A. R. Tackett, G. E. Matthews. A projector augmented wave (PAW) code for electronic structure calculations, Part I: Atompaw for generating atom-centered functions. Comput. Phys. Commun., 135, 329-347(2001).
[71] H. C. Andersen. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys., 72, 2384-2393(1980).
[72] C. Huang, M. Chen, J. Xia et al. Introducing PROFESS 3.0: An advanced program for orbital-free density functional theory molecular dynamics simulations. Comput. Phys. Commun., 190, 228-230(2015).
[73] S. Nosé. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys., 81, 511-519(1984).
[74] W. G. Hoover. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A, 31, 1695-1697(1985).
[75] S. Plimpton. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys., 117, 1-19(1995).
[76] R. Redmer, B. Holst, M. French. Electronic transport coefficients from
[77] R. J. Needs, A. J. Read. Calculation of optical matrix elements with nonlocal pseudopotentials. Phys. Rev. B, 44, 13071-13073(1991).
[78] F. Knider, A. V. Postnikov, J. Hugel.
[79] R. Redmer, M. French. Electronic transport in partially ionized water plasmas. Phys. Plasmas, 24, 092306(2017).
[80] D. R. Hamann. Optimized norm-conserving Vanderbilt pseudopotentials. Phys. Rev. B, 88, 085117(2013).
[81] D. R. Hamann. Erratum: Optimized norm-conserving Vanderbilt pseudopotentials [Phys. Rev. B 88, 085117 (2013)]. Phys. Rev. B, 95, 239906(2017).
[82] A. Dal Corso. Pseudopotentials periodic table: From H to Pu. Comput. Mater. Sci., 95, 337-350(2014).
[83] J. L. Martins, N. Troullier. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B, 43, 1993-2006(1991).
[84] C. E. Wilmer, H. Babaei, P. Boone. Heat flux for many-body interactions: Corrections to lammps. J. Chem. Theory Comput., 15, 5579-5587(2019).
[85] E. A. Carter, C. Huang. Transferable local pseudopotentials for magnesium, aluminum and silicon. Phys. Chem. Chem. Phys., 10, 7109-7120(2008).
[86] C. Meng, J. Yuan, S. Zhang, Y. Hou, C. Gao, D. Kang, J. Zeng. Thermally driven fermi glass states in warm dense matter: Effects on terahertz and direct-current conductivities. Phys. Plasmas, 26, 092701(2019).
[87] B. B. L. Witte, M. French, P. Sperling et al. Observations of non-linear plasmon damping in dense plasmas. Phys. Plasmas, 25, 056901(2018).
[88] A. McKelvey, G. E. Kemp, P. A. Sterne et al. Thermal conductivity measurements of proton-heated warm dense aluminum. Sci. Rep., 7, 7015(2017).

Set citation alerts for the article
Please enter your email address
© Copyright 2018-2021 | Chinese Laser Press. All Rights Reserved 沪ICP备15018463号-20