
Aming LIN, Yiyang SUN. Stability of Low-index Surfaces of Cs2SnI6 Studied by First-principles Calculations [J]. Journal of Inorganic Materials, 2022, 37(6): 691
- Journal of Inorganic Materials
- Vol. 37, Issue 6, 691 (2022)
Abstract
Organic-inorganic hybrid lead halide perovskites, such as CH3NH3PbI3[1⇓-3] have attracted enormous research interests for applications in efficient photovoltaic[4], light- emitting[5] and photodetection devices[6]. As their lead-free counterparts, Sn-based halide perovskites, such as CH3NH3SnI3[7⇓-9] and CsSnI3[10⇓-12], have been proposed for such applications because of their nontoxicity. However, because 2+ state is not the most stable valence state of Sn, the Sn-based perovskites are prone to further oxidation, rendering them even less stable than the Pb-based perovskites, which are already well known to have the stability issue[13]. It is highly desirable to develop air-stable alternatives. For this purpose, Cs2SnI6 is a promising material, in which Sn is already in 4+ valence state and resistant to further oxidation[14⇓-16]. Meanwhile, it has a suitable band gap and strong optical absorption for photovoltaic applications[17⇓-19].
The halide perovskite materials are usually reported to exhibit defect tolerance in the bulk[20-21]. Therefore, the surfaces, interfaces and grain-boundaries are usually the main concerns for optimizing the device performance. The surface properties of Cs2SnI6, which are expected to play an important role in the devices, remain poorly understood. Recently, several groups have devoted to the study on the surfaces of Cs2SnI6. Kapil et al.[22] suggested the existence of the surface state in Cs2SnI6. Shin et al.[23] investigated the role of the surface states in the presence of a redox mediator. Xu et al.[24] reported a general approach to synthesize layered nanoplatelets of Cs2SnI6. Zhu et al.[25] revealed that Cs2SnI6 crystals have a preferential growth of (111) surface. Several experiments also proved that Cs2SnI6 tends to grow along the <111> direction[26⇓-28]. However, it is not clear that this preference is thermodynamically driven or just a result of growth condition specific to individual experiments.
In this work, as motivated by the experimental works, we study the surface properties of Cs2SnI6 using first- principles calculations. As an initiative work, we attempt to understand the preference to the (111) surface in different crystal growth experiments. Different surface models with different surface orientations and terminations were set up to compare their thermodynamic stability. As some terminations are non-stoichiometric, to evaluate their relative stability it is necessary to calculate the chemical potentials to consider the crystal growth conditions. By analyzing the surface stability, it is expected to provide useful information for future experimental synthesis and device fabrication.
1 Computational method
Our first-principles calculations were based on density functional theory and performed using the Vienna Ab initio Simulation Package (VASP)[29]. Projector augmented wave (PAW) potentials were used to describe the interaction between ion cores and valence electrons[30]. The strongly constrained and appropriately normed (SCAN) functional in combination with the rVV10 van der Waals (vdW) functional was used for the exchange-correlation functional[31]. The cutoff energy of planewave basis set was taken to be 340 eV and the Γ-centered 3×3×3 k-point mesh was used for optimizing the 9-atom primitive cell of Cs2SnI6. A cutoff energy of 272 eV and a Γ-centered 3×3×1 k-point mesh were used for the surface calculations.
2 Results and discussion
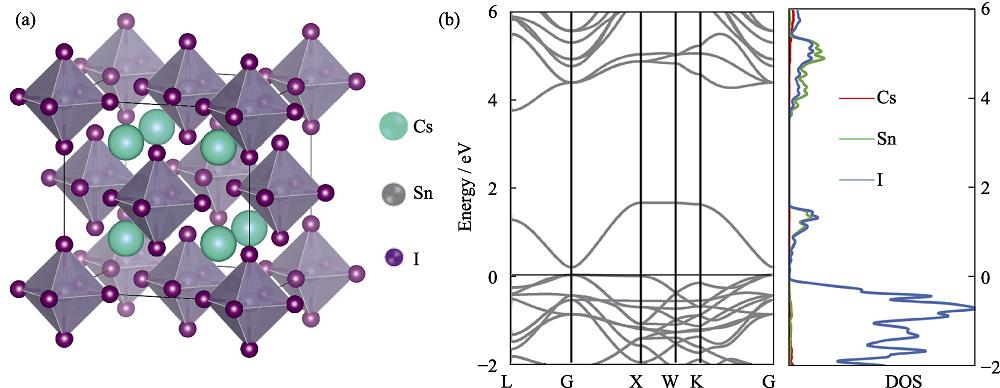
Experimentally, it is reported that bulk Cs2SnI6 exhibits a cubic structure with Fm-3m space group symmetry, as shown in Fig. 1(a), and the lattice parameter a0 is 1.165 nm[20]. The calculated a0 using the SCAN+rVV10 functional is 1.156 nm, 0.8% smaller than the experimental value. The SCAN functional without considering the vdW effect yields a0=1.178 nm, 1.1% larger than the experimental value, suggesting that the vdW effect is significant for Cs2SnI6. For comparison, the commonly used PBE and HSE functional is also considered, which yield a0=1.203 and 1.197 nm, respectively. The reason for this large vdW effect is that the material is rather soft. The calculated bulk modulus using the SCAN+rVV10 method is only 13.1 GPa. Using the other functionals mentioned above would yield even smaller bulk modulus.
![]()
Figure 1.(a) Atomic structure and (b) band structure and projected density of states (pDOS) of Cs2SnI6Colorful figures are available on website
The band structure and projected density of states (pDOS) of Cs2SnI6 were calculated, as shown in Fig. 1(b). The direct gap at the Γ point is 0.19 eV, significantly smaller than the experimental band gap of ~1.3 eV, suggesting that the meta-generalized gradient approximation is not sufficient for studying band-gap-sensitive properties of this material, for which the HSE functional including spin-orbit coupling will be necessary[20]. According to the pDOS plot, the top valence bands are mainly contributed by I5p orbitals, while the bottom conduction bands are contributed by both I5p orbitals and Sn5s orbitals. The Sn5s orbitals form a separate band, above which is another band gap and the Sn5p bands.
The surface properties are studied in the next step. We adopt the symmetric slab models for the surfaces, which possess a mirror symmetry through the middle of the slabs. Such models also avoid spurious interaction between periodic slabs due to dipole-dipole interactions. For all calculations, sufficient vacuum region (more than 1-nm-thick) was used to ensure negligible interaction between the slabs. Seven different terminations of Cs2SnI6 surface models were considered, as shown in Fig. 2. The non-stoichiometric (001) surfaces were modeled with CsI2-terminated (or A-termination) and SnI4-terminated (or B-termination) slabs, whose unit-cell formulae were Cs12Sn5I32 and Cs8Sn5I28, respectively. Similarly, the non-stoichiometric (011) surfaces were modeled with I4-terminated (A-termination) and Cs2SnI2-terminated (B-termination) slabs, whose unit-cell formulae were Cs10Sn5I34 and Cs10Sn5I26, respectively. Along the [111] direction, the atomic stacking sequence is -Sn-CsI3- CsI3- Sn-. Correspondingly, the non-stoichiometric Sn-terminated (A-termination), non- stoichiometric CsI3-terminated (B-termination) and stoichiometric CsI3-terminated surfaces were modeled, whose unit-cell formulae were Cs8Sn5I24, Cs12Sn5I26 and Cs10Sn5I30, respectively.
![]()
Figure 2.Seven supercell models of Cs2SnI6 surfaces (a) For (001) surface: CsI2-terminated and SnI4-terminated slabs; (b) For (011) surface: I4-terminated and Cs2SnI2-terminated slabs; (c) For (111) surface: non-stoichiometric Sn-terminated, CsI3-terminated and stoichiometric CsI3-terminated slabs
The cleavage energy are firstly evaluated, which is the energy required to split a crystal into two complementary non-stoichiometric terminations. It is noted that CsI2- and SnI4-terminations for Cs2SnI6 (001) surfaces are mutually complementary, and so are I4- and Cs2SnI2-terminated slabs for (011) surfaces, as well as Sn- and CsI3-terminated slabs for (111) surfaces. As two complementary surfaces (also referred to as A- and B-termination above) are created simultaneously when a crystal is cleaved, the total cleavage energy of two complementary surfaces can be obtained by
where $E(\mathrm{~A})_{\text {slab }}^{\text {unrel }} \text { and } E(\mathrm{~B})_{\text {slab }}^{\text {unrel }}$ are the energies of unrelaxed A- and B-terminated slabs, respectively.Ebulk is the energy per unit cell, S represents the surface area and n is the total number of bulk unit cells in the two slabs. Next, the relaxation energy of A-terminated surfaces with both sides relaxed is calculated according to
where $E(\mathrm{~A})_{\text {slab }}^{\mathrm{rel}}$ is the energy of A-terminated slab after relaxation.Erel(B) is calculated similarly. Finally, the total surface energy of the two complementary surfaces can be obtained as the sum of the cleavage and relaxation energies:
The calculated results of total cleavage energy, total relaxation energy and total surface energy of the two complementary non-stoichiometric terminations with different surface orientations are shown in Fig. 3. For comparison, the cleavage, relaxation and surface energies of the stoichiometric CsI3-terminated (111) surface are also shown. It can be seen that the total surface energies of the two complementary non-stoichiometric terminations are relatively high compared with that of stoichiometric CsI3-terminated (111) surface whose surface energy is only 0.11 J/m2, regardless of the surface orientations. However, the contributions to the cleavage energy from the A- and B-terminations are not equal. Further study is needed to determine whether A- or B-termination could individually have surface energy lower than 0.11 J/m2. In order to evaluate the relative stability of each surface termination under various experimentally preparation conditions, the consideration of chemical potential μCs, μSn and μI is necessary[21,32].
![]()
Figure 3.Calculated total cleavage, relaxation and surface energies of two complementary non-stoichiometric terminations in (001), (011) and (111) orientations, which are compared with the cleavage, relaxation and surface energies of the stoichiometric CsI3-terminated (111) surface
Through varying the chemical potentials, different experimental conditions can be simulated. The relative values of the chemical potentials of Cs, Sn and I are introduced with respect to their most stable phases, $\Delta \mu_{\mathrm{Cs}}=\mu_{\mathrm{Cs}}-E_{\mathrm{Cs}}^{\text {bulk }}, \Delta \mu_{\mathrm{Sn}}=\mu_{\mathrm{Sn}}-E_{\mathrm{Sn}}^{\text {bulk }}$, and $\Delta \mu_{\mathrm{I}}=\mu_{\mathrm{I}}-\frac{1}{2} E_{\mathrm{I}_{2}}^{\text {bulk }}$. The chemical potentials of Cs, Sn and I are constrained by the calculated enthalpy of formation of the primary phase Cs2SnI6[33-34]
where $\Delta E_{\mathrm{f}}\left(\mathrm{Cs}_{2} \operatorname{SnI}_{6}\right)$ is calculated to be -9.16 eV. The chemical potentials are further subject to specific bounds that are set by the existence of secondary phases:
The enthalpies of formation above were all obtained by SCAN+rVV10 calculations. By considering these constraints, the final allowed region for μSn and μI is demarcated by the points A, B, C, D and E, where equilibrium growth of bulk Cs2SnI6 is possible, as shown in Fig. 4.
![]()
Figure 4.Illustration of the accessible chemical potential region for Cs2SnI6Constraints imposed by the formation of competing secondary phases resulting in the allowed region shaded in green
Using the determined chemical potential region, the surface energy for each individual termination can be obtained using[35-36]
where Eslab is the total energy of relaxed A-termination, NCs, NSn and NI are the numbers of Cs, Sn and I atoms in the slab, respectively. Considering the variation of chemical potential with reference phase as mentioned above, the surface energy can be finally rewritten as
Here, Ø(A) is a constant term, independent of the chemical potentials. Considering that surfaces will spontaneously form and the crystal will be destroyed if surface energy is negative, it is necessary to satisfyEsurf>0.
The stability diagram of the Cs2SnI6 (001) surface is shown in Fig. 5(a). The blue and orange regions represent the regions where CsI2- and SnI4-terminations are stable, respectively. The upper part of the green region is located in the blue region, indicating that the CsI2-termination is favored under the I-poor condition. There is still a small part of the green region located in the orange region, e.g., at chemical potential points C and D, indicating that the SnI4-termination is more stable than the CsI2-termination under I-rich condition.
![]()
Figure 5.Stability of low-index surfaces of Cs2SnI6 as a function of chemical potentials (a) Analysis of stability of the two terminations of Cs2SnI6 (001) surface with respect to the allowed region for maintaining equilibrium with the primary phase Cs2SnI6. The orange and blue regions indicate the stable region for CsI2- and SnI4-terminations, respectively; (b) Similar to (a) for the Cs2SnI6 (011) surface. The orange and blue regions are for the I4- and Cs2SnI2-terminations, respectively; (c) Similar to (a) for the Cs2SnI6 (111) surface. The orange and blue regions are for the Sn- and stoichiometric CsI3-terminations, respectively; (d) Surface energies of the seven surface models of Cs2SnI6 as a function of the chemical potentials colorful figures are available on website
Similarly, the stability diagram of the Cs2SnI6 (011) surface is shown in Fig. 5(b). The blue region represents that the Cs2SnI2-termination is thermodynamically more stable, while the orange part refers to the region where the I4-termination is more stable. It can be seen that the green region is also located in both blue and orange regions, indicating that different terminations are favored when varying the experimental environments. Under I-rich condition (e.g., at chemical potential point A) the Cs2SnI2-termination is favored, while under I-poor condition (e.g., the chemical point D) the I4-termination is favored.
In Fig. 5(c), the stability diagram of Cs2SnI6 (111) surface is shown. Different from the stability diagrams of the (001) and (011) surfaces discussed above, the whole green region is located in the orange region for the (111) surface, indicating that the stoichiometric CsI3-terminated (111) surface is the most energetically favored among the three terminations regardless of the chemical potentials.
Finally, the surface energies of the seven terminations of Cs2SnI6 low-index surfaces are compared in Fig. 5(d) as a function of the chemical potentials. It can be seen that the stoichiometric CsI3-terminated (111) surface consistently has the lowest surface energy, indicating that it is the most thermodynamically favored surface among the seven terminations, in agreement with the recent experimental reports[25-26].
3 Conclusion
Based on density-functional theory calculations with the SCAN+rVV10 functional, seven models for the low- index Cs2SnI6 surfaces were studied with different surface orientations and terminations to compare their thermodynamic stability. Overall, based on the calculated surface energies, we identified that the stoichiometric CsI3-termination for (111) surface is consistently the most stable, regardless of the chemical potentials, which is in agreement with the experimental observation that the (111) surface is often the most exposed surface. For the (100) and (110) surfaces, two different terminations were considered for each of them. Their relative stability depends on the chemical potentials. From an experimental point of view, when preparing these two surfaces, different terminations can be obtained by varying the growth condition, e.g., by controlling the I-poor or I-rich conditions.
Acknowledgement
The authors thank Professor Lian Jie, Dr. Zhu Weiguang and Shen Junhua from Rensselaer Polytechnic Institute for enlightening discussions.
References
[23] H O SHIN, B M KIM, T JANG et al. Surface state-mediated charge transfer of Cs2SnI6 and its application in dye-sensitized solar cells. Advanced Energy Materials, 18032(2019).
[24] Y XU, S LI, Z ZHANG et al. Ligand-mediated synthesis of colloidal Cs2SnI6 three-dimensional nanocrystals and two-dimensional nanoplatelets. Nanotechnology, 2956(2019).
[25] W ZHU, J SHEN, M LI et al. Kinetically controlled growth of sub-millimeter 2D Cs2SnI6 nanosheets at the liquid-liquid interface. Small, 2006(2021).


Set citation alerts for the article
Please enter your email address

© Copyright 2018-2021 | Chinese Laser Press. All Rights Reserved 沪ICP备15018463号-20