
Guoping Luo, Yingmei Bian, Ruifeng Wu, Guoxia Lai, Xiangfu Xu, Weiwei Zhang, Xingyuan Chen. First principles study of the electronic structure and photovoltaic properties of β-CuGaO2 with MBJ + U approach[J]. Journal of Semiconductors, 2020, 41(10): 102102
- Journal of Semiconductors
- Vol. 41, Issue 10, 102102 (2020)
Abstract
1. Introduction
Oxide semiconductors generally have large band gaps, which can be widely used in thin-film transistors of the ultraviolet region, light-emitting diodes or transparent electrodes, etc.[
2. Theoretical model and calculated method
The calculations in this paper are carried out by the Vienna ab-initio simulation package (VASP) software package based on the density functional theory (DFT) theory[
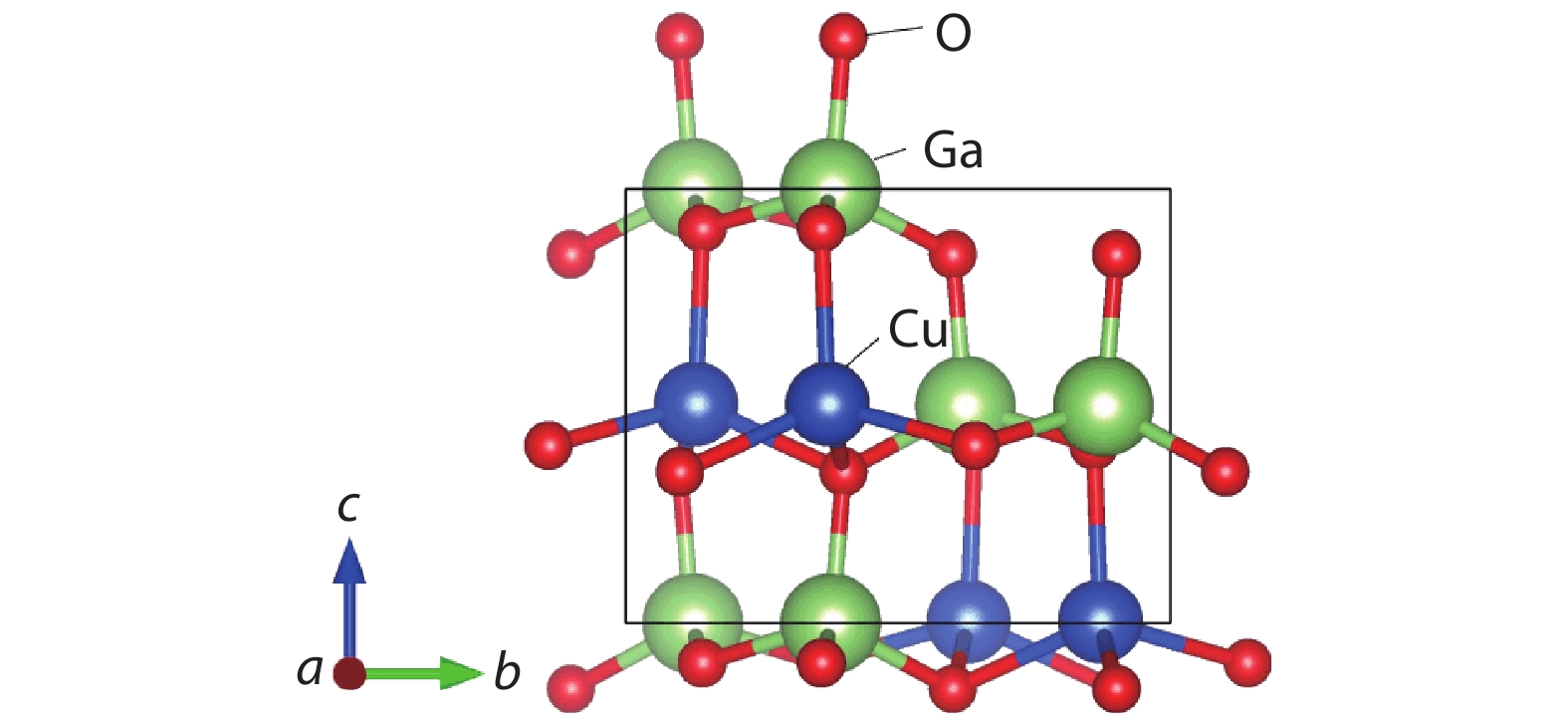
![]()
Figure 1.(Color online) The structure of the
3. The calculated results and discussion
3.1. The band structure and electronic structure
As shown in Fig. 2, the calculated band gap value does not exceed 1 eV by GGA + U with the values of U changing from 4 to 10 eV, which is deviated from the experimental value of 1.47 eV. However, in the calculations of MBJ + U approach, the calculated band gap value is very close to the experimental value when U takes 7 and 8 eV. Finally, we select MBJ + U (U = 7.3 eV) approach, and the calculate band gap value of β-CuGaO2 is 1.46 eV. In the calculation of the latter electronic structure, we all take the parameters of MBJ + U (U = 7.3 eV), which ensures the reliability of our calculation.
![]()
Figure 2.(Color online) The band gap values of
As shown in Fig. 3, the valence-band maximum (VBM) and the conduction band bottom (CBM) of β-CuGaO2 are both at the G point, which indicated a direct band gap material. The curve near VBM is relatively flat, indicating that the effective mass of the hole is relatively large, while the effective mass of electrons is relatively small since the curve near CBM is steep, which is consistent with the early reported results[
![]()
Figure 3.(Color online) The band structure and DOS of
3.2. Optical properties
The absorption coefficients [α(E)] of β-CuGaO2 were obtained by the following formula:
where c is the speed of light in vacuum, ω is the incident light frequency. The imaginary and real parts of the dielectric function are expressed in terms of εi and εr, respectively. Fig. 4 shows the calculated absorption coefficients spectrum of β-CuGaO2. The AM 1.5 solar spectral irradiance in the 300–1000 nm wavelength range is also shown in Fig. 4. It can see that β-CuGaO2 exhibit high absorption coefficient up to 105 cm–1 in the visible region. The total fractional absorption (A) of incident solar radiation can be approximately expressed as following formula with the film thickness (d) and absorption coefficient [α(E)].
![]()
Figure 4.(Color online) The absorption coefficient of
where Eg is the band gap and SE is the incident solar spectral irradiance changing with incident photon energy E. As film thickness of β-CuGaO2 changes, the calculated fraction absorption of incident solar flux is shown in Fig. 5. As the thickness of β-CuGaO2 thin films increases from 0.1 to 1 μm, the total fractional absorption slightly increased, and then starts to saturate with thickness of 3 μm.
![]()
Figure 5.(Color online) The optical absorption properties of
3.3. Photovoltaic properties
The Shockley-Queisser limit indicates an absorber with bandgap nearby 1.4 eV has higher photovoltaic conversion efficiency[
![]()
Figure 6.(Color online) The SLME
![]()
Figure 7.(Color online) The current density–voltage (
4. Conclusions
In summary, GGA + U and MBJ + U approach with lower calculated costs are employed to calculate the electronic structure of β-CuGaO2 in this paper. It is found that the calculated band gap value obtained by MBJ + U (U = 7.3 eV) approach is closer to the experimental value. Near the top of the valence band of β-CuGaO2 is mainly composed of the Cu-3d energy level and the O-2p energy level. The bond formed by the hybridization between Cu-3d energy level and the O-2p energy level determines the main properties of the material. When the thickness of β-CuGaO2 film is increased from 0.1 to 1 μm, the total absorption of the film increases sharply. The total absorption becomes to saturation when the film thickness is about 3 μm. The photovoltaic performance parameters of idea β-CuGaO2 solar cells are calculated by the SLME method, which indicated that it could reach 32.4% energy conversion efficiency. By comparison with photovoltaic parameters of CdTe thin film solar cells, it was found that β-CuGaO2 could be a potential high-efficiency photovoltaic material.
Acknowledgements
This work was supported by the NSFC (Grant No. 11547201), Natural Science Foundation of Guangdong Province, China (Grant No. 2017A030307008), Natural Science Basic Research Program of Shaanxi (Program No. 2019JQ-380), and Natural Science Foundation of Guangdong Petrochemical University of Technology, China (Grant No. 2017rc20).
References
[1] K Ellmer. Past achievements and future challenges in the development of optically transparent electrodes. Nat Photonics, 6, 809(2012).
[2] T Minami. Transparent conducting oxide semiconductors for transparent electrodes. Semicond Sci Tech, 20, S35(2005).
[3] C Klingshirn. The luminescence of ZnO under high one- and two-quantum excitation. Phys Status Solidi B, 71, 547(1975).
[4] H Tang, K Prasad, R Sanjines et al. Electrical and optical properties of TiO2 anatase thin films. J Appl Phys, 75, 2042(1994).
[5] P W Baumeister. Optical absorption of cuprous oxide. Phys Rev, 121, 359(1961).
[6] T Omata, H Nagatani, I Suzuki et al. Wurtzite-derived ternary I–III–O2 semiconductors. Sci Tech Adv Mater, 16, 024902(2015).
[7] T Omata, H Nagatani, I Suzuki et al. Wurtzite CuGaO2: A new direct and narrow band gap oxide semiconductor applicable as a solar cell absorber. J Am Chem Soc, 136, 3378(2014).
[8] S Song, D Kim, H M Jang et al.
[9] C N Berglund, H J Braun. Optical absorption in single-domain ferroelectric barium titanate. Phys Rev, 164, 790(1967).
[10] W Ji, K Yao, Y C Liang. Bulk photovoltaic effect at visible wavelength in epitaxial ferroelectric BiFeO3 thin films. Adv Mater, 22, 1763(2010).
[11] H Okumura, K Sato, T Kakeshita. Electronic structure, defect formation energy, and photovoltaic properties of wurtzite-derived CuGaO2. J Appl Phys, 123, 161584(2018).
[12] L Wang, T Maxisch, G Ceder. Oxidation energies of transition metal oxides within the GGA +
[13] I Suzuki, H Nagatani, M Kita et al. First principles calculations of ternary wurtzite
[14] J Heyd, G E Scuseria, M Ernzerhof. Hybrid functionals based on a screened Coulomb potential. J Chem Phys, 118, 8207(2003).
[15] M Shishkin, G Kresse. Implementation and performance of the frequency-dependent GW method within the PAW framework. Phys Rev B, 74, 035101(2006).
[16] J Hafner. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J Comput Chem, 29, 2044(2008).
[17] L Yu, A Zunger. Identification of potential photovoltaic absorbers based on first-principles spectroscopic screening of materials. Phys Rev Lett, 108, 068701(2012).
[18] G Kresse, J Furthmüller. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput Mater Sci, 6, 15(1996).
[19] G Kresse, D Joubert. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B, 59, 1758(1999).
[20] J P Perdew, K Burke, M Ernzerhof. Generalized gradient approximation made simple. Phys Rev Lett, 77, 3865(1996).
[21] A I Liechtenstein, V I Anisimov, J Zaanen. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys Rev B, 52, R5467(1995).
[22] A D Becke, E R Johnson. A simple effective potential for exchange. J Chem Phys, 124, 221101(2006).
[23] F Tran, P Blaha. Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys Rev Lett, 102, 226401(2009).
[24] Y Zhang, Y Wang, L Xi et al. Electronic structure of antifluorite Cu2X (X = S, Se, Te) within the modified Becke-Johnson potential plus an on-site Coulomb
[25] W Shockley, H J Queisser. Detailed balance limit of efficiency of p-n junction solar cells. J Appl Phys, 32, 510(1961).
[26] X Huang, T R Paudel, S Dong et al. Hexagonal rare-earth manganites as promising photovoltaics and light polarizers. Phys Rev B, 92, 125201(2015).
[27] M A Green, K Emery, Y Hishikawa et al. Solar cell efficiency tables (Version 45). Prog Photovolt: Res Appl, 23, 1(2015).

Set citation alerts for the article
Please enter your email address
© Copyright 2018-2021 | Chinese Laser Press. All Rights Reserved 沪ICP备15018463号-20