1State Key Laboratory on Integrated Optoelectronics, College of Electronic Science and Engineering, Jilin University, Changchun 130012, China
2State Key Laboratory of High Field Laser Physics, Shanghai Institute of Optics and Fine Mechanics, Chinese Academy of Sciences, Shanghai 201800, China
3Centre d’Optique, Photonique et Laser et le Département de Physique, Université Laval, Quebec City, Quebec G1V0A6, Canada
Huailiang Xu, Ruxin Li, See Leang Chin. Photoemission mechanisms of methane in intense laser fields[J]. Chinese Optics Letters, 2015, 13(7): 070007
Copy Citation Text
The visible and near-UV emission spectroscopy of methane () induced by a femtosecond intense laser field (800 nm, 40 fs, ) is studied. By measuring the decay profiles of the neutral fragment product CH (), two reaction pathways, i.e., the electron-ion recombination through and the direct disintegration of are found to be responsible for populating the electronic excited states of the neutral fragment product CH, which gives rise to the photoemissions. Our results provide complementary information on previous understanding of the strong-field-induced photoemission mechanism of through neutral dissociation of superexcited states.
The interaction of molecules with femtosecond intense laser fields has, both experimentally and theoretically, been a subject of intense interest in recent years[1,2]. In particular, filamentation induced by the propagation of femtosecond laser pulses in air can produce a strong field of over a long distance that enables molecules in the propagation path of filaments[3] to be remotely ionized/dissociated. This opens up the possibility of investigating strong-field molecule interactions at a remote place in a variety of complicated atmospheric environments[4]. However, because of the difficulty to remotely characterize molecular reaction dynamics using ions or electron signals, photoemission signals are usually employed to explore related phenomena[5–8].
Photoemission signals induced by intense laser fields in filaments have been observed from a variety of gaseous molecules including hydrocarbons[9,10], halocarbons[11], carbon dioxide[12], and major atmospheric constituents: nitrogen[5,8,13], oxygen[14], and water vapor[6,7,15]. The mechanisms behind the photoemission of gaseous molecules in intense laser fields have also been extensively studied. For example, the optical emission from ( transition) was ascribed to direct ionization of inner-valence electrons of neutral nitrogen molecules[16], and that of ( transition) was attributed either to the two indirect excitations by the reactions of followed by in the case of higher electron density[17] and the collision-assisted intersystem crossing from excited singlet states in the case of lower electron density[18], or to the direct excitation by electron impact with a circularly polarized pulse[19].
Hydrocarbon molecules such as , , and exhibit identical optical emissions but remarkably different signal intensities from neutral fragments of CH (, , , (), and in a femtosecond laser filament[9,10,20]. However, the underlying mechanism responsible for the photoemission phenomenon of the neutral fragments is still not totally clear. Only one mechanism has been so far proposed, which attributed the strong-field-induced photoemission phenomenon to neutral dissociation of parent hydrocarbon molecules via superexcited states (SESs), i.e., states beyond the ionization limit[21].
Sign up for Chinese Optics Letters TOC. Get the latest issue of Chinese Optics Letters delivered right to you!Sign up now
In the present study, we systematically investigate the photoemission from the transition of the free radical CH induced from by a femtosecond laser field at the intensity of about in order to examine whether or not there exist other contributions to the photoemission of hydrocarbon molecules in intense laser fields. By using time-resolved measurements, two reaction pathways, i.e., the electron-ion recombination of and the direct decomposition of ions, are unambiguously identified to be responsible for the CH emission of in intense laser fields.
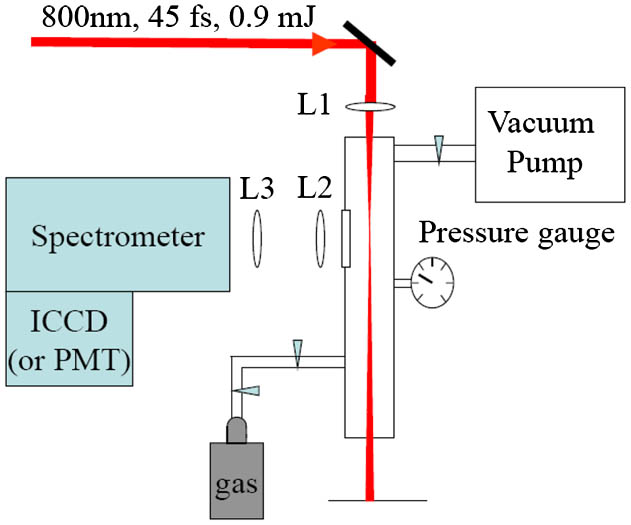
The experiment was conducted using a Ti:Sapphire femtosecond laser system. The pulses, emitted from a Ti:Sapphire oscillator (Spectra Physics Tsunami), were positively chirped to about 200 ps in a stretcher. After this, the pulses with a repetition rate of 1 kHz were successively amplified in a regenerative amplifier and a two-pass Ti:Sapphire amplifier (Spectra Physics Spitfire). A compressor was used to shorten the pulse duration, measured with a single-shot autocorrelator (SSA Positive Light), to 45 fs at FWHM. The pulse spectrum was centered at 800 nm with a 23-nm bandwidth (FWHM). The diameter of the beam was about 5 mm (at level of intensity) and the energy per pulse was about 0.9 mJ. As shown in Fig. 1, the laser beam was focused by a fused silica lens (L1: ) into a vacuum chamber filled with at a pressure range of 0.1–40 Torr. The thickness of the input fused silica window was about 6 mm and the distance between the focusing lens and the input window of the vacuum chamber was about 2 cm. The induced fluorescence signal was collected at a right angle to the laser propagation direction. The fluorescence passed through an 8-cm diameter fused silica window and was imaged by two convex quartz lenses (L2: and L3: ) onto the entrance slit of the imaging spectrometer (Acton Research Corp., Spectrapro-500i). The spectral resolution of the spectrometer was about 0.4 nm using a grating of (blazed wavelength at 500 nm) with a 100 μm entrance slit width. The entrance slit of the spectrometer was arranged to be parallel to the laser propagation direction in order to enhance the collection efficiency. The dispersed fluorescence was detected using a gated intensified charge coupled device (ICCD, Princeton Instruments PI-Max: 512) or using a Hamamatsu R5916U-52 microchannel-plate photomultiplier tube with a rise time of 0.2 ns. A Tektronix Model TDS 7254 oscilloscope with a bandwidth of 2.5 GHz was used to record and average the signals from the photomultiplier tube.
Figure 2 demonstrates the femtosecond laser-induced fluorescence spectra of in the spectral region of 300–700 nm for the pressures of 0.1, 1, and 10 Torr, respectively. The ICCD gate width was set to 1 μs and the ICCD delay time was set to (note that the laser pulse arrives in the interaction region at ). The data were averaged over 200 shots. The spectral lines were assigned to CH (, , ), (Swan band) and H ()[9]. It can be noted that in Fig. 2 the Swan band of can only be clearly observed in the spectrum for a pressure of 10 Torr or more. Since there is only one carbon atom existing in the parent molecule, the collisions among the induced fragments of ionization products could be one of the mechanisms for the product of [22]. As for CH and H, they have been previously attributed to the neutral dissociation of through the SESs by a pump-probe experiment in which a fast depletion of the CH fluorescence signal with a lifetime of about 160 fs was observed as a function of the delay time between a pump (800 nm) pulse and a probe (1338 nm) pulse[23]. The 800-nm pulse prepares the molecule in the SESs, and the probe pulse de-excites the SESs, leading to a decrease of the CH signal produced through neutral dissociation of SESs of [23]. However, it should be pointed out that the decrease in the fluorescence signal of CH was only 5%[23], which indicates that the dissociation of the SESs of might not be the only mechanism responsible for the product of the CH fragment. Therefore, we investigate in the following the decay profiles of the transition of CH at 431 nm using time-resolved measurements in order to determine whether there exist other mechanisms contributing to the photoemission of CH.
Figure 2.Femtosecond laser-induced emission spectra of : 0.1 Torr (bottom), 1 Torr (middle), and 10 Torr (top). The ICCD gate width was set to 500 ns and the ICCD delay time (note that the laser pulse arrives in the interaction region at ).
Figure 3 shows a typical decay profile of the transition of CH at 431 nm. The pressure is 0.2 Torr (black line). The data were averaged over 100 laser shots. The decay profile can be well fitted to three exponentials (red line) with the time constants of , , and . One of these three exponential decays can be assigned to the spontaneous decay of the excited state of CH. As already indicated in the pump-probe experiment[23], the neutral dissociation of via its SESs is one source of populating the excited state. However, this decay process is very fast, occurring on a femtosecond time scale, and thus can be interpreted as being the ‘immediate’ population of the excited state, which emits spontaneously. As a result, the contribution from the SESs of in the decay profile should show a sharp rise time, which is limited by the detector system’s rise time, and then a decrease with the spontaneous decay constant of the state. Therefore, besides the neutral dissociation of via its SESs, it should have two other channels to make contributions to the excited state of CH.
Figure 3.Decay profile (black line) of the fluorescence emission at 431.2 nm from the transition of CH for the pressures at 0.2 Torr and its fit (red line). The insets show the same data but in a semilogarithmic representation.
Next, we attempt to explore the possible mechanisms responsible for the product of CH lying in the state. Since three exponentials are needed to fit the decay of the state, the decay dynamics of the state should be described by a rate equation with three exponential terms. Therefore, a rate equation including the terms due to electron-ion recombination and disintegration of is written as[17]where and are the spontaneous decay rate and the population of the excited state, respectively; and refer to the reaction rates of the electron-ion recombination and the disintegration of processes; and refers to the population/density of species . This rate equation is based on the fact that the main positive component in the plasma is according to the mass spectra of [24] and the laser intensity used in this work (). That is, the reaction channels from other positive ions such as (the second main positive ion in the plasma[24]) can be neglected. Otherwise, more than three exponentials due to additional terms in the equation will be obtained. Here it is reasonable to assume that and [electron] decay exponentially with a rate of and , respectively. Finally, the solution of , where is a constant used to fit the experimental data. As a result, we measure the decay curves of the fluorescence emission at 431 nm at different pressures and fit them by Eq. (2). It is found that all of the experimental data can be well fitted, with uncertainties of less than 2%. By plotting the decay rates (inverse lifetimes) as a function of pressure, the lifetime extrapolated at a pressure of is about 530 ns (see Fig. 4), which agrees very well with the previously measured radiative lifetime of the excited state of CH (about 537 ns[25]). This corresponds to the value of at a pressure of . Therefore, the values of () and measured at a pressure of can be assigned to the decay times of electrons and in the plasma, respectively.
The agreement between the present and previous lifetimes of the state of CH indicates the reasonability of the written rate equation. In addition, because of the large difference between and , these two processes could not be due to the same type of reactions, i.e., both due to the electron-ion recombination of or due to the disintegration of multiply charged ions . As a consequence, besides the neutral dissociation of SESs, as proposed previously[20,23,26], the photoemission from the transition of CH can also result from the electron-ion recombination of and the disintegration of .
In conclusion, we study the emission spectroscopy of induced by intense laser fields. In order to understand the mechanisms responsible for the photoemission of CH, we measure the emission spectra of , the decay curves, and the decay rate as a function of pressure. Two possible reaction channels are unambiguously identified for producing the photoemission of CH: one is the recombination of electrons and ions by and the other is the disintegration of . When combined with the previously confirmed contribution from the neutral dissociation of SESs of , our results provide complementary information on understanding the mechanism for the CH emission of in intense laser fields. In view of the complexity of ionization, fragmentations, and collisions of molecules and molecular ions in strong laser fields[27–29], all of which may affect the induced photoemissions, it is clear that additional effort is needed to give a complete understanding of photoemissions of molecules in intense laser fields.