
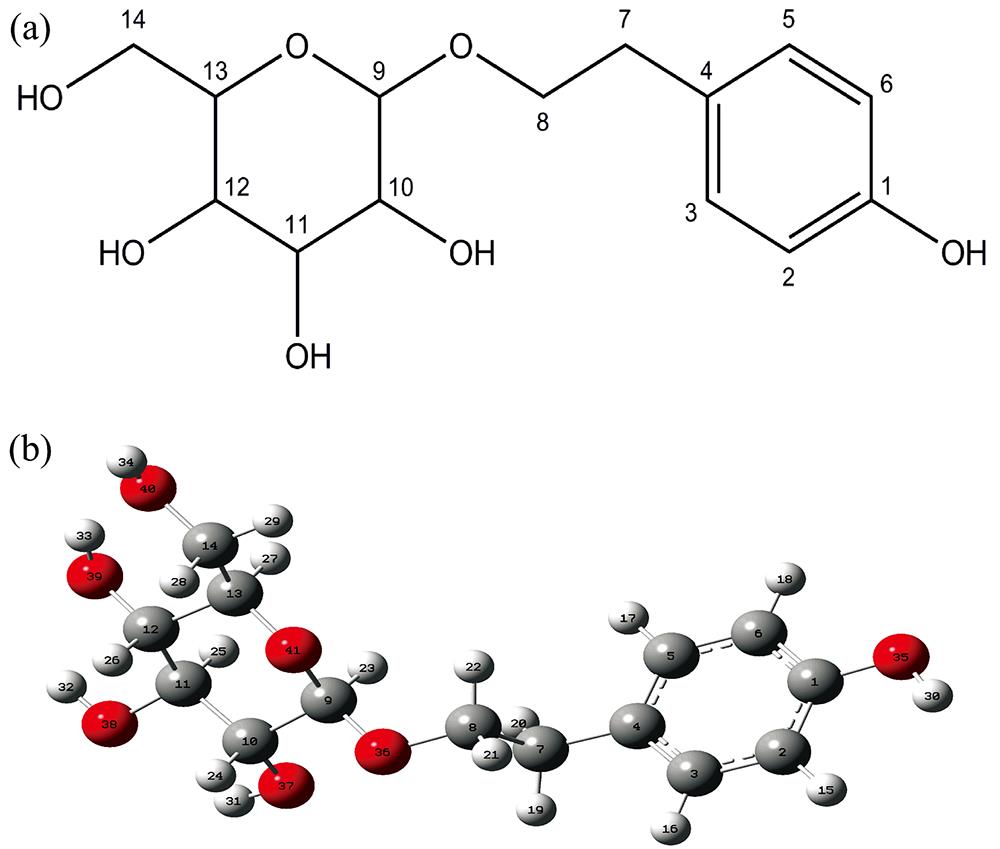
Yu-yu XIE, Xue-ling HOU, Zhi-hui CHEN, Haji Akber AISA. Density Functional Theory Studies on Structure and Spectra of Salidroside Molecule[J]. Spectroscopy and Spectral Analysis, 2022, 42(6): 1786
- Spectroscopy and Spectral Analysis
- Vol. 42, Issue 6, 1786 (2022)
Abstract

Set citation alerts for the article
Please enter your email address
© Copyright 2018-2021 | Chinese Laser Press. All Rights Reserved 沪ICP备15018463号-20