Understanding the photophysical interactions between the components in organic-inorganic nanocomposites is a key factor for their efficient application in optoelectronic devices. In particular, the photophysical study of nanocomposites based on organic conjugated polymers is rare. We investigated the effect of surface plasmon resonance (SPR) of gold nanoparticles (Au NPs) on the photoluminescence (PL) property of a push-pull conjugated polymer (PBDB-T). We prepared the hybrid system by incorporating poly(3-hexylthiophene)-stabilized Au NPs (P3HT-Au NPs) into PBDB-T. The enhanced and blueshifted PL was observed in the hybrid system compared to PL in a neat PBDB-T system, indicating that the P3HT chains attached to the Au NPs suppressed charge-transfer from PBDB-T to the Au NPs and relayed the hot electrons to PBDB-T (the band-filling effect). This photophysical phenomenon limited the auto-dissociation of PBDB-T excitons. Thus, the radiative recombination of the excitons occurred more in our hybrid system than in the neat system.
1. INTRODUCTION
Hybrid nanomaterials of semiconductors (SCs) and plasmonic metal nanoparticles (NPs) [1–9] such as gold (Au) and silver have received considerable attention in the field of optoelectronic devices because of their unique photophysical properties [10–37]. The energy flow control from surface plasmon resonance (SPR) of the plasmonic NPs to the SCs is a critical factor to increase the energy conversion efficiency in the plasmonic hybrid system [13,18,23,25,33]. This factor can induce hot-electron injection (HEI) [18,19,22] and plasmon-induced resonance energy transfer (PIRET) [24,25,27]. The suppression of the singlet () exciton quenching from the SCs to the NPs should be considered for efficient HEI and PIRET [38,39].
The spectral overlap, distance, and energy-level arrangement between the SCs and the plasmonic NPs have important effects on the design of the hybrid systems with more efficient HEI and PIRET [13–16]. In particular, these photophysical phenomena heavily rely on the distance between the components in the hybrid systems [27,30]. Thus, plasmonic hybrid systems based on conjugated polymers (CPs) deserve significant attention because they can be prepared through various nanostructure-fabrication methods [40–43]. The organic CP-based optoelectronic devices can be facilely fabricated in a large area by low cost solution processing [44]. Despite these advantages, the photophysical phenomena in the CP-based plasmonic hybrid system are not extensively investigated.
Recently, we reported the effect of HEI induced by poly(3-hexylthiophene)-stabilized Au NPs (P3HT-Au NPs) on the excited-state dynamics of the organic matrix [18]. To more efficiently achieve the HEI effect on the optical property of the hybrid system than the previous report, we designed the plasmonic hybrid system by incorporating P3HT-Au NPs into a push-pull-type CP matrix [PBDB-T, Fig. 1(a)]. For the clear clarification of the SPR effect on PBDB-T -excitons, we prepared the hybrid films with the PBDB-T/P3HT-Au NPs blend ratios of 3:1 and 1:1 (mass ratio). We expected that the -exciton quenching from the PBDB-T matrix to the Au NPs would be more suppressed than that in the previously reported hybrid system containing the P3HT matrix because the conduction band energy-level of PBDB-T is lower than that of P3HT [45–48].
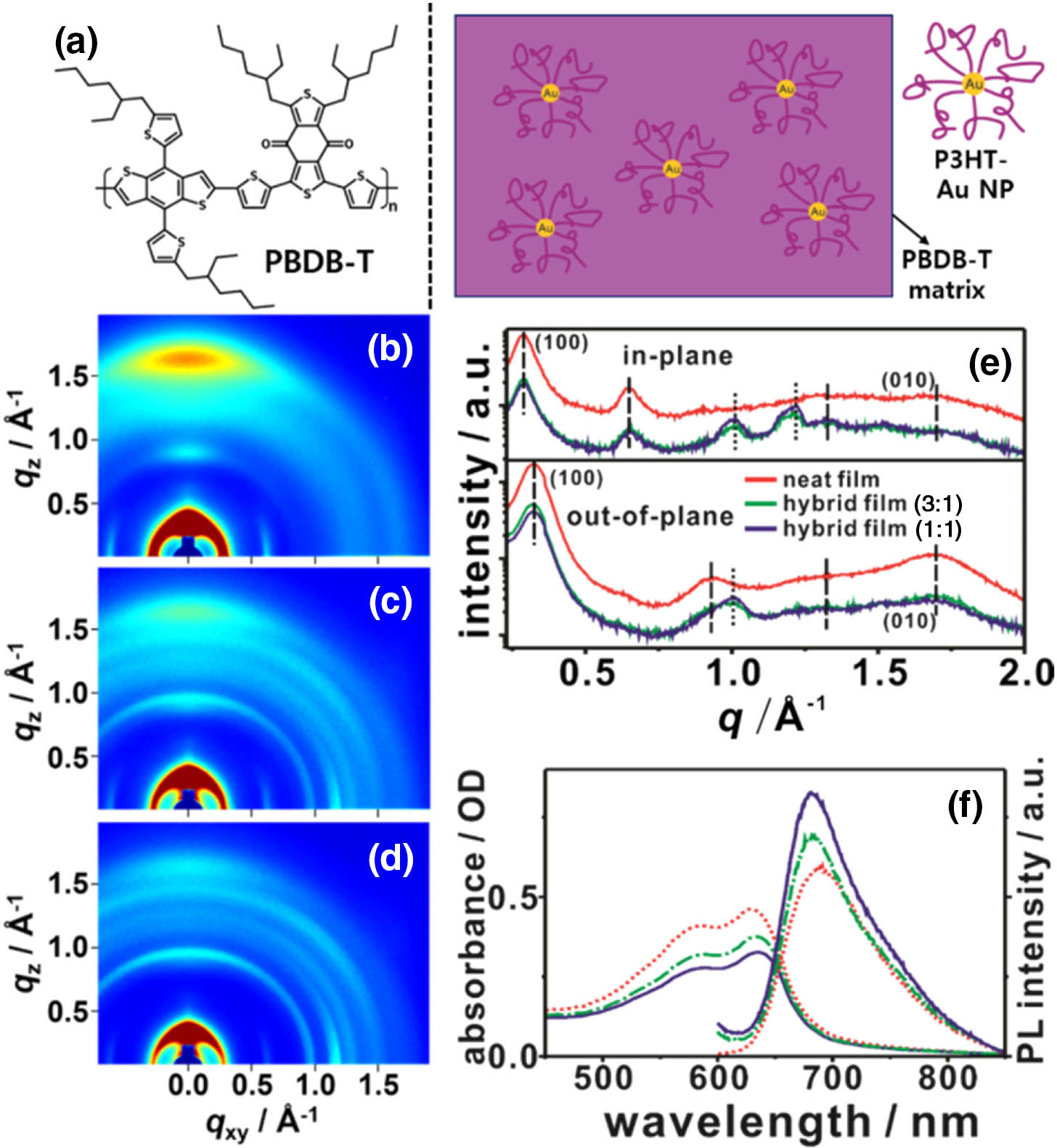
Figure 1.(a) Left, chemical structure of PBDB-T and, right, a schematic representing the plasmonic hybrid system of a PBDB-T matrix and P3HT-Au NPs. 2D GI-XRD patterns of (b) a neat PBDB-T film and hybrid films with PBDB-T/P3HT-Au NPs mass ratios of (c) 3:1 and (d) 1:1. (e) (out-of-plane) and (in-plane) scans obtained from the GI-XRD patterns. (f) Left, absorption and, right, PL spectra of the neat film (dotted) and the hybrid films with the ratios of 3:1 (dotted–dashed) and 1:1 (solid). The samples were excited at 520 nm to obtain the PL spectra.
Poly[(2,6-(4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione))](PBDB-T) was purchased from 1-Material Inc. P3HT-Au NPs were synthesized by reducing an Au precursor directly with a propylthiol-terminated P3HT (P3HT-SH) surfactant, as described in detail elsewhere (the specification of THT P3HT-SH used in this study; , PDI = 1.28, MALDI-MS , , and degree of polymerization = 40) [38]. The gravimetric ratio of inorganic Au NPs to organic P3HT is 3:1, and the grafting density of P3HT-SH chains attached to Au NP is approximately 9 P3HT chains/Au NP, [41]. 10 mg of P3HT-Au NPs or PBDB-T was dissolved in 1 mL of chlorobenzene (CB). We prepared the plasmonic hybrid films with the CB solution volume-ratios of 3:1 and 1:1 (PBDB-T/P3HT-Au NPs); 0.1 mL or 0.3 mL of the P3HT-Au NPs CB solution was added into 0.3 mL of the PBDB-T CB solution. The homogeneous blend-solutions of P3HT-Au NPs and PBDB-T were spin-coated on glass plates at 1000 r/min for 60 s. Thus, the content ratios of Au NPs, the P3HT-SH chains attached to Au NPs, and the PBDB-T matrix in our hybrid system are 18.7:6.30:75.0 and 37.5:12.5:50.0 (mass ratio).
B. Characterization
UV-visible absorption spectra were measured using a Perkin Elmer Lambda 1050 spectrometer, and photoluminescence (PL) spectra were measured using a Horiba Jobin Yvon NanoLog Spectrofluorometer. Transmission electron microscopy (TEM) images were obtained by using a JEOL JEM-1011. A high resolution (HR) TEM image was obtained by using a JEOL JEM-2200FS equipped with Image Cs-corrector. 2D grazing incidence X-ray diffraction (GI-XRD) experiments were performed using the synchrotron source at the Pohang Accelerator Laboratory. The refractive indices of the thin films for the PL quantum yields (PLQYs) were obtained using a Woollam spectroscopic ellipsometer (M2000D). For the femtosecond transient absorption (fsTA) data of the films, an Ultrafast Systems HELIOS-femtosecond transient absorption spectrometer and a Libra 1 kHz Femtosecond Ti:sapphire Regenerative Amplifier system with a center wavelength of 780 nm and an 80 fs pulse duration were used [18].
C. Finite-Difference Time-Domain Simulation
We used the finite-difference time-domain (FDTD) modules within the Lumerical DEVICE Suite to obtain the PL enhancement factor of the hybrid system. The FDTD simulation is a classical theory method based on Maxwell’s equations [49]. To mimic the complex decay mechanisms in the neat and hybrid systems, we assumed that the dipole source is all radiation light that can be generated from the systems. The ratios of radiative electric field power (, neat system; , hybrid system) were regarded as the ratios of radiative decay rates (, neat system; , hybrid system) [49]. We started the FDTD simulation assuming the neat PBDB-T system was in the form of a homogeneous cube. The volume-fraction ratio of Au NPs to the PBDB-T matrix is about 1:26; the Au NP diameter was assumed to be 5 nm, and the PBDB-T density is about [50]. The P3HT chains attached to Au NPs were excluded in this simulation. We obtained the refractive indices of PBDB-T and the Au NPs from an ellipsometry measurement and the literature, respectively [51]. The distance between the dipole source and the localized surface plasmon (LSP) of the Au NPs was fixed to 1.5 nm.
To obtain the initial electric field power (), the dipole source was surrounded with six power monitors. We obtained the electric field power emitted from the PBDB-T matrix (, without Au NPs; , with Au NPs) by surrounding the total system with the power monitors and summarizing each monitored power. We set the mesh step with 0.5 nm of the mesh override regions covering the total system. After the simulation for the hybrid system is completed, we removed the Au NPs from the hybrid system, and the resulting void was refilled with the PBDB-T cube; the simulation for the neat system ran with the same configuration on the hybrid system. From the collected simulation data, we obtained the PL enhancement factor of the hybrid system compared to the neat system ().
D. Kinetic Analysis
For global and target analyses of the obtained fsTA data, we performed singular value decomposition (SVD) using MATLAB and Surface Xplorer software. The transient absorption (TA) data can be decomposed into the sum of exponential decays as where is the decay-associated difference spectra (DADS), is the kinetic constant, and is the number of decay component. The target analysis can be conducted according to the following equation: where is the species-associated spectra (SAS), is the number of species, and is the species concentration. We used the nonlinear kinetic equations of Eq. (3) for the target analysis,
3. RESULTS AND DISCUSSION
2D GI-XRD patterns and 1D XRD profiles of the thin films in Figs. 1(b)–1(e) show the incorporation-effect of P3HT-Au NPs on the nanostructure of our plasmonic hybrid system. The reflection peaks in the GI-XRD pattern and profiles of the PBDB-T neat film are in good agreement with those in the previous reports [Figs. 1(b) and 1(e), dashed lines] [52]. The (010) reflection with a -spacing of 3.71 Å (the π-π stacking distance; the interchain ordering of PBDB-T, 1 Å = 0.1 nm) on the axis ( Å-1) is much more intense than that on the axis, indicating that a face-on orientation dominantly exists more than an edge-on orientation in the neat film [52]. The reflection peaks around Å on all axes in the GI-XRD images and profiles of the hybrid films [Figs. 1(c)–1(e),dotted lines] originated from the intrachain aggregation of the P3HT chains attached to the Au NPs [41]. The weakened intensity and the broad widths of the (010) reflection peaks on both and axes in the XRD profiles of the hybrid films compared to those in the XRD profiles of the neat PBDB-T film are clearly observed, whereas the widths of the (100) reflection peaks on both axes in the XRD profiles (in-plane, 0.29 Å-1; out-of-plane, 0.32 Å-1) of the hybrid films are similar to those on both axes in the XRD profiles of the neat film [Fig. 1(e)]. This result indicates that the crystallinity of the PBDB-T matrix decreases compared to that of the neat PBDB-T film; in particular, the disordering of the PBDB-T interchain-packing in the hybrid films increased mainly.
Absorption spectrum of the neat film [left in Fig. 1(f), dotted line] has two peaks at 580 and 620 nm that are assigned to the 0-1 and 0-0 transitions of aggregated PBDB-T chains, respectively [53–55]. These peaks are also clearly observed in the absorption spectra of the hybrid films [left in Fig. 1(f), dotted–dashed and solid lines]. Figure 2 indicates that the broadbands below 550 nm slightly increased in the absorption spectra of the hybrid films compared to that in the absorption spectra of the neat PBDB-T film, indicating that the Au NPs interrupted the crystallization of the PBDB-T chains in the hybrid films [39]. In addition, the peak-intensity ratios of (0-0)/(0-1) in the absorption spectra of the hybrid films are larger than that in the absorption spectrum of the neat film (Fig. 2), which indicates that the 0-0 transition associated with the intrachain-ordering of the PBDB-T chains is more dominant than the 0-1 transition associated with the interchain-packing of the PBDB-T chains in the hybrid film [41,53–55]. This result well matches the XRD result in Fig. 1(e). However, although the crystallinity of the PBDB-T matrix in the hybrid films decreased compared to that of the neat PBDB-T film, the main absorption peaks in the absorption spectra of the hybrid films were slightly redshifted from those in the absorption spectrum of the neat film (Fig. 2); the incorporation of the P3HT-Au NPs into the PBDB-T matrix can result in two types of PBDB-T domains (the disordered PBDB-T domains with the P3HT-Au NPs and the bulk PBDB-T ordered domains without the P3HT-Au NPs). Thus, we suggest that the phase separation between the disordered domains and the ordered domains caused the redshift of main absorption peaks in the absorption spectra of the hybrid films compared to those in the absorption spectrum of the neat PBDB-T film.
Figure 2.Peak-normalized absorption spectra of a neat PBDB-T film (dotted) and hybrid films with PBDB-T/P3HT-Au NPs mass ratios of 3:1 (dotted–dashed) and 1:1 (solid).
In particular, although the optical density (OD) of the redshifted main peaks in the absorption spectra of the hybrid films is lower than that in the absorption spectrum of the neat film, a main emissive peak at 690 nm in a PL spectrum of the neat film [dotted, right in Fig. 1(f)] was gradually enhanced [, right in Fig. 1(f)] and blueshifted in the PL spectra of the hybrid films with increasing P3HT-Au NP content. When preparing the hybrid films, since PBDB-T and the P3HT-Au NPs were separately dissolved in CB and then blended, the total amount of CB solvent was inevitably increased compared to that when preparing the neat PBDB-T film. Therefore, the hybrid films were made thinner than the neat film, so the OD values of absorption spectra of the hybrid system were lower than that of absorption spectrum of neat PBDB-T system. We obtained the PL enhancement factors of the hybrid films by dividing the PL intensities [dotted–dashed and solid in Fig. 1(f)] of the hybrid films by the PL intensity [dotted in Fig. 1(f)] of the neat PBDB-T film; the PL enhancement factor at 670 nm is 1.22 for the hybrid film with the ratio of 3:1 and 1.48 for the hybrid film with the ratio of 1:1. We also obtained the PLQYs of the thin films, which are listed in Table 1.
Many studies have been reported to introduce the plasmonic NPs into the active layers, to improve the power conversion efficiencies of organic-polymer solar cells by the localized surface plasmon resonance (LSPR) effects of plasmonic metal NPs on optical properties of active layers; this strategy enhanced optical absorption in the active layers [56–58]. In case of our hybrid system, the incorporation of the P3HT-stabilized Au NPs into the PBDB-T matrix can result in two factors that may affect the optical property of the hybrid system: the structural change of the PBDB-T matrix and the interaction between singlet () excitons of PBDB-T and SPR of Au NPs. If the structural change is more dominant than the photophysical interaction in the hybrid system, the Stokes shifts of the hybrid films should be larger than that of the neat PBDB-T film due to the larger structural relaxation of the amorphous PBDB-T chains in the disordered domains with the P3HT-Au NPs following photoexcitation [59]. However, the Stokes shifts of hybrid films are smaller than that of neat PBDB-T film [Figs. 1(f) and 2]. Thus, the PL result strongly implies that the photophysical interaction between PBDB-T and SPR of the Au NPs exists in our hybrid system, which induced the radiative recombination of PBDB-T -excitons or the increase in the -exciton concentration.
To elucidate the PL enhancement phenomenon in the hybrid system compared to the neat system, we calculated a PL enhancement factor of the hybrid system through an FDTD method (Fig. 3). Our FDTD calculation focused on the wavelength range from 650 nm to 710 nm where the PL intensity of the hybrid system clearly increased compared to that of the neat system. As mentioned earlier, the experimental PL enhancement factor [Fig. 3(a)] was obtained by dividing the PL intensity of the hybrid film with the 1:1 ratio by the PL intensity of the neat film.
Figure 3.PL enhancement factor of the hybrid film with the 1:1 ratio compared to the neat PBDB-T film: (a) the experimental value and (b) the simulation value obtained by a finite-difference time-domain (FDTD) method.
The FDTD method has the limitations to quantify excitation and non-radiative decay rates [49]. Thus, we excluded the excitation and non-radiative processes of the both systems in this simulation, and the total radiative decay rate of the both systems was fixed to the same value . We obtained the simulated PL enhancement factor [Fig. 3(b)] using the following equations [49]: where and represent the radiative decay rate of the neat PBDB-T film and the hybrid film with the 1:1 ratio, respectively, and represents the electric field power emitted from the PBDB-T matrix (, without Au NPs; , with Au NPs). The ratio of that corresponds to the radiative decay ratio () can be regarded as the PL enhancement factor in the hybrid system compared to the neat system; the relative PL enhancement was indirectly determined by simulating the electronic field power emitted from the both systems (with or without the SPR effect of the Au NPs).
The simulated PL enhancement factor by the SPR effect on the radiative decay rate in the hybrid system is clearly seen at 670 nm, which is in good agreement with the experimental result; the maximum experimental PL enhancement factor is also observed at 670 nm. We confirmed the loss of the electric field power due to the Au NPs in this simulation. Despite this loss, the radiative decay process more occurs in the hybrid system than in the neat system. From the FDTD simulation result, we anticipate that the excited electrons are more generated in the hybrid system due to SPR of the Au NPs than in the neat system.
The PL enhancement factor at 670 nm is 2.12 for the simulation value and 1.48 for the experimental value; the reason for the difference between the experimental and theoretical values is explained as follows. Unlike the experimental PL enhancement result in the hybrid system, our FDTD simulation cannot completely reproduce the randomly dispersed P3HT-Au NPs in the PBDB-T matrix, so the simulation is performed by assuming the periodic shape of a local space where the Au NPs are evenly distributed. Therefore, the change in PL intensity is sensitive to slight adjustments to the Au NP position within the polymer matrix. If the Au NP position is repeatedly adjusted to bring the theoretical value closer to the experimental value, the difference can be reduced. However, we did the FDTD simulation to clearly confirm the PL enhancement phenomenon with the presence or absence of the SPR effect of Au NPs in the overall system, rather than matching the experimental and theoretical values. Thus, we omitted the process of repeatedly adjusting the position of Au NPs in the PBDB-T matrix. To more clarify the PL enhancement mechanism in the hybrid system, we obtained fsTA data of the thin films.
The TA spectra of the neat film have one broad photoinduced absorption (PIA) peak at 1180 nm that clearly splits into two peaks after 430 fs [Fig. 4(a), 900 and 1180 nm]. For precise assignment of these PIA peaks, we obtained fsTA spectra of the blend film of PBDB-T and an acceptor molecule (). The PIA peak at 1180 nm in the TA spectrum of the blend film was drastically decreased within 1 ps compared to that in the TA spectrum of the neat film, while the PIA peak at 900 nm shows the opposite trend (Fig. 5). Thus, we assigned the PIA peaks at 900 and 1180 nm to the positive polaron () and -exciton species of PBDB-T, respectively [60].
Figure 4.Transient absorption (TA) spectra of (a) a neat PBDB-T film and hybrid films with PBDB-T/P3HT-Au NPs mass ratios of (b) 3:1 and (c) 1:1.
Because a nonlinear process, e.g., singlet-singlet annihilation, may interrupt the clear analysis of the SPR effect of the Au NPs on the PBDB-T -excitons in our hybrid system, we obtained the TA data of the thin films with a pump intensity of μ to confirm the excited-state dynamics in a linear-decay regime; the TA kinetics with low pump intensities (those under μ) decayed similarly (Fig. 6 and Table 2).
Figure 6.Pump-intensity-dependent TA kinetics of the neat PBDB-T film pumped with 620 nm and probed at 1180 nm. Dotted lines are best-fitted curves to extract decay constants.
The polaron peak at 900 nm was not well observed in the TA spectra of the hybrid films in Figs. 4(b), 4(c), and 7(a), indicating that the incorporation of the P3HT-Au NPs into the PBDB-T matrix suppressed the formation of the species in the hybrid films. In particular, the -excitons in the hybrid films have much slower relaxation and recombination dynamics than those in the neat film [Fig. 7(b) and Table 3]. This result indicates that the charge-transfer from PBDB-T to the Au NPs occurred rarely due to the P3HT chains attached to the Au NPs [18]. With increasing P3HT-Au NP content in the hybrid system, these tendencies are clearly observed. In the bottom of Fig. 7, we schematically described the excited-state dynamics in the hybrid system changed by the incorporation of the P3HT-Au NPs into the PBDB-T matrix; the first is the suppressed polaron formation, and the second is the slowdown in -exciton decays.
Figure 7.(a) Transient-absorption (TA) spectra and (b) kinetics of the neat PBDB-T film and the hybrid films. The samples were excited at 520 nm of μ and probed at 1180 nm for the singlet ()-exciton TA kinetics of the films. The schematic kinetic-diagrams representing the excited-state dynamics of the neat and the hybrid systems (bottom): i, the relaxation process; ii, the formation of positive () and negative polarons (); iii, the geminate recombination of -excitons to the ground state.
DADS [Figs. 8(a)–8(c)] obtained from a global analysis of the fsTA data of the films with Eq. (1) clearly show that the hybrid film with the ratio of 1:1 has the slowest decay-dynamics among our prepared films. The suppressed polaron-formation dynamics in the hybrid system compared to the neat system are clearly seen in the DADS of Fig. 8. For the neat film [Fig. 8(a)], the -exciton peak at 1180 nm is clearly seen in the first two DADS (squares: , 378 fs; circles: , 3.86 ps), almost disappears from the third and fourth DADS (triangles: , 39.6 ps; inverted triangles: , inf.), and is replaced by the polaron peak at 900 nm. The presence of the -exciton peak is more observable in the third decay component of the hybrid film with the ratio of 3:1 [Fig. 8(b); , 98 ps] than in the third decay component of the neat film [Fig. 8(a); , 40 ps]. In particular, the maximum OD value of the -exciton peak is larger than that of the polaron peak in the second decay-component of the hybrid film with the ratio of 1:1 [Fig. 8(c); , 39 ps].
Figure 8.Decay-associated difference spectra obtained from the TA data fitted globally to an exponential decay equation described in Eq. (1): (a) the neat film; (b) the hybrid film with the ratio of 3:1; (c) the hybrid film with the ratio of 1:1.
The two types of PBDB-T domains (the disordered PBDB-T domains with the P3HT-Au NPs and the bulk PBDB-T ordered domains without the P3HT-Au NPs) were formed within the hybrid films by adding the P3HT-Au NPs to the PBDB-T matrix, which resulted in the hybrid films with the lower crystallinity compared to the neat film (see the XRD and absorption spectral results in Fig. 1). Thus, the relaxation process of PBDB-T -excitons is expected to proceed faster in the hybrid films than in the neat PBDB-T film because the energy transfer from the disordered domains to the ordered crystalline domains occurs well more in the hybrid films than in the neat PBDB-T film [61,62]. However, our TA results clearly show the opposite tendency: the slowdown in the relaxation process in the hybrid films compared to the neat film. This is reliable evidence that the SPR of the Au NPs has a much more important effect on the excited-state dynamics of the hybrid system than the structural change caused by introducing the P3HT-Au NPs to PBDB-T; the photophysical interaction between PBDB-T and the Au NPs disturbs the energy transfer from the disordered PBDB-T domains to the ordered crystalline PBDB-T domains.
To investigate the effect of the structural change on the polaron-formation dynamics in the hybrid films, we obtained molecular-weight () dependent absorption spectra (Fig. 9)and DADS (Fig. 10) of PBDB-T: the high film with 80–100,000 g/mol and the low film with 40,000 g/mol. The 0-0 transition is more dominant than the 0-1 transition in the neat film of high PBDB-T, whereas the opposite tendency is observed in the neat film of low PBDB-T (Fig. 9) [55]. This indicates that the intrachain-ordering of PBDB-chains is more dominant than the interchain-packing of PBDB-T chains in the high neat film. The dependent DADS (Fig. 10) show that the polarons were generated well more in the high neat film than in the low neat film. In the high neat film [Fig. 10(a)], the -exciton PIA peak at 1180 nm is clearly seen in the first decay component (squares; , 860 fs). It almost disappeared from the second decay component (circles; , 21.9 ps), and was replaced by the polaron PIA peak at 900 nm. On the other hand, in case of the low neat film [Fig. 10(b)], both the -exciton PIA peak at 1180 nm and the polaron PIA peak at 900 nm are well observed in the second decay component (circles; , 29.9 ps). Roy et al. [63] reported that the backbone planarization in push-pull-type CPs leads to intramolecular charge-transfer (ICT), facilitating efficient charge-carrier generation: . Thus, the polarons were well more generated through ICT induced by the backbone planarization character of the intrachain-ordering in the high neat film than in the low neat film; the interchain-packing of PBDB-T chains disturbs the formation of localized ICT state.
Figure 9.Absorption spectra of the neat PBDB-T films.
Figure 10.Decay-associated difference spectra obtained from the TA data of the neat films fitted globally to an exponential decay equation described in Eq. (1).
In case of our hybrid system, the incorporation of the P3HT-Au NPs into the PBDB-T matrix induced the partial disordering of the PBDB-T interchain-packing (the 0-1 transition) in the hybrid films, resulting in the intrachain-ordering effect being more increased than the interchain-packing effect in the hybrid films (Fig. 2). This structural change is expected to promote the polaron formation in the hybrid systems. However, the suppressed polaron formation in the hybrid system compared to those in the neat system is clearly observed in our TA results (Figs. 7 and 8). Thus, we insist that the changed excited-state dynamics in the hybrid system compared to that in the neat system are due to the photophysical interaction between the Au NPs and the PBDB-T -excitons rather than the structural changes that are caused by the incorporation of the P3HT-Au NPs into PBDB-T.
To more clearly demonstrate the effect of the photophysical interaction between SPR of the Au NPs and the PBDB-T -excitons on the polaron dynamics in the hybrid system, we conducted a target analysis of the fsTA data with Eq. (2) (Fig. 11); we used the simplified kinetic-model that includes the nongeminate recombination dynamics of the polaron species (scheme 1), where and represent the ground state and singlet states, respectively, and and are free polarons. SAS [Figs. 11(a) and 11(b)] and model population kinetics [Figs. 11(c) and 11(d)] of the -states and the species in the films clearly indicate the effect of the P3HT-Au NPs on the excited-state dynamics of the PBDB-T matrix: the suppressed polaron formation and the slowdown in the -exciton decays. The TA kinetics of the selected wavelengths fitted well to the Eq. (3) are shown in Figs. 11(e) and 11(f). The time constants obtained from this analysis are listed in Table 4. The polaron ()-formation dynamics (, ) are much slower in the hybrid film with the ratio of 1:1 (3.31 ps) than in the neat film (0.742 ps). The slowdown in the recombination dynamics of the -exciton and the species is also observed in the hybrid system. The free polarons recombine nongeminately to the ground state; they have the concentration dependent decay-dynamics. Thus, the much larger decay-constant of the hybrid film (38.9 ps) than that of the neat film (14.5 ps) clearly indicates the suppressed polaron formation in the hybrid film. The model population kinetics of the given species [Figs. 11(c) and 11(d)] are in good agreement with the SAS results. The population ratio of (solid) drastically decreased with the incorporation of the P3HT-Au NPs into the CP matrix.
Figure 11.(a), (b) Species-associated spectra and (c), (d) model population kinetics of the given species. (e), (f) The selective wavelengths kinetics of the TA data fitted globally to the nonlinear kinetic equations described in Eq. (3). [(a), (c), and (e) the neat film; (b), (d), and (f) the hybrid film with the 1:1 ratio.]
Recently, we reported the slowdown in the excited-state dynamics of the hybrid system containing the P3HT-Au NPs, which is induced by HEI from the Au NPs to the P3HT matrix [18]. The P3HT chains attached to the Au NP play two critical roles in the hybrid system based on the P3HT matrix, as in this study; the spacer role is in suppressing the exciton quenching loss of the P3HT matrix to Au NPs, and the bridge role is to relay the hot electrons to the P3HT matrix. The injected hot electrons in the P3HT chains attached to Au NPs can be delocalized to the P3HT matrix, resulting in the flow of the excess energy from the Au NPs to the P3HT matrix [18]. Thus, HEI induced the slowdown in the excited-state dynamics of the hybrid system based on the P3HT matrix compared to that of the neat P3HT system. However, because the matrix in the hybrid system is also P3HT, it is not possible to completely suppress the back charge-transfer from the P3HT matrix to the Au NPs; the enhanced PL property is not observed in the PL spectrum of the hybrid film compared to that in the PL spectrum of the neat P3HT film [18].
We incorporated the P3HT-Au NPs into the push-pull conjugated polymer (PBDB-T) matrix to more efficiently suppress the back charge-transfer from the CP matrix to the Au NPs; we expected that the P3HT chains attached to the Au NPs that have the higher conduction band energy-level than PBDB-T efficiently prevented the -exciton quenching loss of the PBDB-T matrix in the hybrid system [45–48]. In spite of the redshifted main peaks in the absorption spectra of the hybrid films (Fig. 2), the blueshifted PL property of the hybrid system compared to the neat system [Fig. 1(f)] strongly implies that energy from SPR of the Au NPs flows into the PBDB-T matrix; the Stokes shifts of hybrid films are smaller than that of neat PBDB-T film. In addition, we confirmed the PLQY of the thin films and the PL enhancement factor of the hybrid film with the FDTD method (Table 1 and Fig. 3). These results clearly indicate that our energy-alignment strategy caused the efficient SPR effect of the Au NPs on the organic CP matrix more than that reported previously. The optical-density values of -excitons in the hybrid films are similar to that in the neat PBDB-T film in the femtosecond time window (see Fig. 4), indicating that PIRET from the Au NPs to PBDB-T occurred rarely in the hybrid films. Thus, we suggest the PL enhancement mechanism induced by HEI from the Au NPs to PBDB-T in the hybrid system (Fig. 12).
Figure 12.Schematic representing the excited-state dynamics of the neat PBDB-T system (left) and schematic representing the changed excited-state dynamics of the hybrid system by the hot-electron injection (HEI) from the P3HT-coated Au NPs to the PBDB-T matrix (right; the HEI-induced PL enhancement mechanism and the suppression of back charge-transfer from PBDB-T to the Au NPs by the P3HT chains tethered to the Au NPs).
We describe in the following the HEI effect on the excited-state dynamics of PBDB-T that induced the enhanced and blueshifted PL property of the hybrid system. Upon excitation, hot electrons with higher energy than the Schottky barrier were generated via the non-radiative decay process of a plasmon [13]. The charge-transfer from the P3HT chains attached to the Au NPs occurred in the P3HT-Au NPs, resulting in the empty conduction band of P3HT. The P3HT -quenching loss mechanism caused by the charge-transfer from P3HT to the Au NPs at the interface has been reported [39,64]. It results in the empty conduction band of P3HT [64].
Figure 13 indicates the P3HT -quenching loss mechanism in the P3HT-Au NPs. The PIA peak around 1300 nm (the -excitons) in the TA spectrum of the P3HT-Au NPs film (squares) was drastically decreased within 1 ps compared to that in the TA spectrum of the neat P3HT film (circles), while the PIA peak around 1000 nm (the positive polarons, ) shows the opposite trend. We also confirmed the effect of energy transfer from the P3HT chains attached to the Au NPs to the PBDB-T matrix on the photophysical property of the hybrid system (Fig. 14).
Figure 13.TA spectra of the neat P3HT and P3HT-Au NPs films. The samples were excited at 520 nm.
We prepared the blend film of P3HT/PBDB-T and compared the PL property to that of the neat PBDB-T film; the enhancement of PL intensity is not observed in the blend film of P3HT/PBDB-T (dotted in Fig. 14) compared to the neat PBDB-T film (solid in Fig. 14). If the energy transfer from P3HT to PBDB-T occurs, the PL peak around 700 nm in the PL spectrum of the blend film of P3HT/PBDB-T should be intensified compared to that in the PL spectrum of the neat PBDB-T film. The TA (Fig. 13) and PL (Fig. 14) spectra of the thin films indicate that because the P3HT chains within our hybrid system are tethered to the Au NPs, the charge-transfer from P3HT to the Au NPs is much more dominant than the energy transfer from P3HT to PBDB-T in our plasmonic hybrid system. Thus, we suggest that the energy transfer from P3HT to PBDB-T rarely affected the PL enhancement mechanism of our plasmonic hybrid system.
The hot electrons with higher energy than the Schottky barrier can be injected into the empty conduction band of the P3HT chains attached to the Au NPs due to the efficient P3HT -quenching loss within the P3HT-Au NPs. The conduction band energy-level of PBDB-T is lower than that of P3HT [45,47]. Thus, the hot electrons with excess energy in the P3HT chains were readily transferred into the PBDB-T matrix (the energy-delocalization), which can raise the PBDB-T -states to the higher energy state and fill the lowest energy-level in the conduction band of the PBDB-T matrix (the band-filling effect). This photophysical interaction between the hot electrons and PBDB-T resulted in the slowdown in the relaxation-dynamics and the blueshifted PL property of the hybrid films compared to that of the neat film [Fig. 1(f)] [65].
The polaron formation occurs in competition with the -exciton relaxation, which can be explained by the hot-exciton dissociation model [66]. In case of our hybrid system, the energy-delocalization between the PBDB-T -excitons and the hot electrons in the P3HT chains occurred competitively with the polaron formation. HEI from the Au NPs to PBDB-T occurred more efficiently than the ICT process; thereby, the auto-dissociation of PBDB-T -excitons reduced. As a result, the radiative recombination of the PBDB-T -excitons more occurred in our hybrid system than in the neat system. In addition, as mentioned earlier, the P3HT spacer that has the higher conduction band energy-level than PBDB-T efficiently prevented the back charge-transfer from PBDB-T to the Au NPs (the -exciton quenching loss of the PBDB-T matrix) in the hybrid system. Thus, we suggest that the PL enhancement mechanism in our hybrid system is due to HEI from the Au NPs to the PBDB-T matrix (Fig. 12).
4. CONCLUSION
In conclusion, we elucidated the HEI-induced PL enhancement mechanism in the hybrid system of P3HT-Au NPs and a push-pull-type CP (PBDB-T) using the FDTD simulation and transient absorption spectroscopy. The P3HT chains attached to the Au NPs efficiently suppressed the exciton quenching loss from PBDB-T to the Au NPs and relayed excess energy of the hot electrons from the Au NPs to PBDB-T, which induced the slowdown in the -exciton dynamics and the suppression of the polaron formation in our hybrid system. This photophysical interaction resulted in the enhanced and blueshifted PL property in our hybrid system compared to that in the neat system. Thus, our findings deserve attention in the field of the plasmonic hybrid optoelectronic devices.
Acknowledgment
Acknowledgment. The authors thank the Pohang Accelerator Laboratory (PAL) for providing the synchrotron radiation sources at 9A beamline.