
Wenyu Fang, Wenbin Kang, Jun Zhao, Pengcheng Zhang. Surface-regulated triangular borophene as Dirac-like materials from density functional calculation investigation[J]. Chinese Physics B, 2020, 29(9):
- Chinese Physics B
- Vol. 29, Issue 9, (2020)
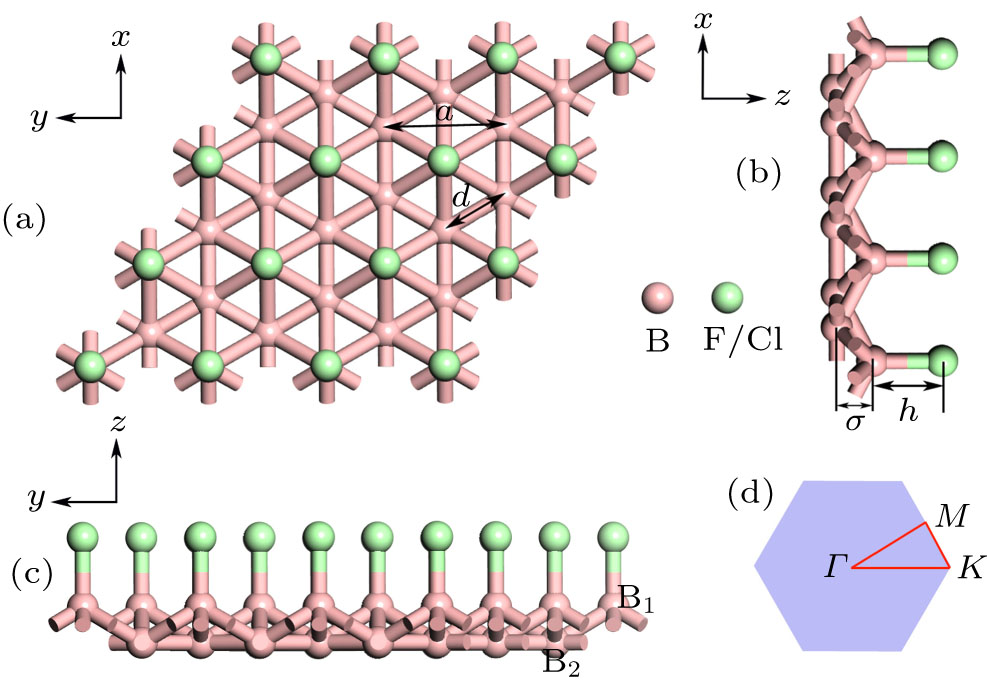
Fig. 1. The optimized geometric structures for the B3X (X = F, Cl) and the 2D Brillouin zone (BZ) of the monolayers exhibiting high symmetry k -points.
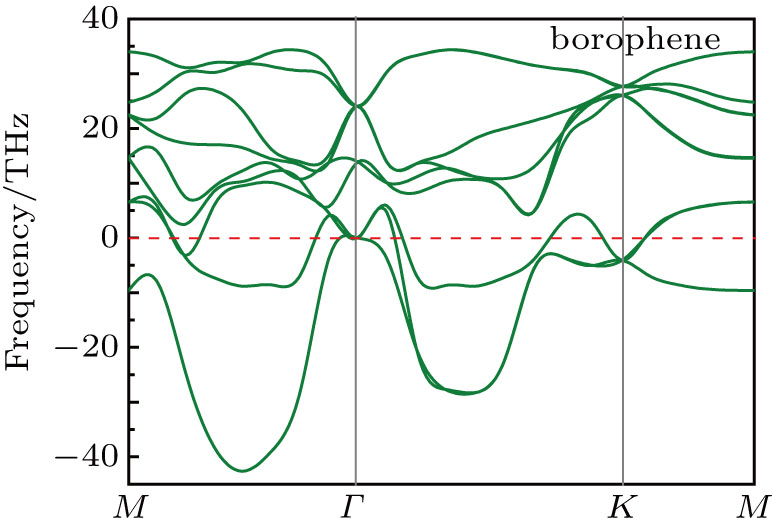
Fig. 2. Phonon dispersion of triangular borophene.
Fig. 3. Phonon dispersion of (a) B3F and (b) B3Cl.
Fig. 4. Variation of the total energy in the molecular dynamics simulation at 500 K during a timescale of 10 ps for (a) B3F and (b) B3Cl. The insets give the top (left panel) and side (right panel) views of the atomic structure snapshots taken from the molecular dynamics simulation.
Fig. 5. Polar diagram for (a) Young’s modulus and (b) Poisson’s ratio of B3X (X = F, Cl). θ is the angle with respect to the a -direction.
Fig. 6. Electronic band structures and density of states of (a) B3F, (b) B3Cl, (c) B3Br, and (d) B3I.
Fig. 7. Calculated in-plane and out-plane light absorption coefficients of monolayer B3F and B3Cl, using the screened HSE06 hybrid functional.
|
Table 1. Calculated lattice constant a, bond lengths d, h, buckling height σ, and binding energy Ef (marked in Fig. 1 ) as well as electronic energy band gaps (based on both PBE and HSE06 functionals) of all chemically decorated triangular borophene.
|
Table 2. The elastic constants of all chemically decorated triangular borophene calculated by GGA + PBE.
| |||||||||||||||||||
Table 3. Fermi velocities at the valence band top of B3F and B3Cl (based on both PBE and HSE06 functionals).


Set citation alerts for the article
Please enter your email address

© Copyright 2018-2021 | Chinese Laser Press. All Rights Reserved 沪ICP备15018463号-20