
Juefei Wu, Wang Yue-Chao, Yu Liu, Bo Sun, Yanhong Zhao, Jiawei Xian, Xingyu Gao, Haifeng Liu, Haifeng Song. First-principles study on the electronic structure transition of β-UH3 under high pressure[J]. Matter and Radiation at Extremes, 2022, 7(5): 058402
- Matter and Radiation at Extremes
- Vol. 7, Issue 5, 058402 (2022)
Abstract
I. INTRODUCTION
Hydrogen-rich materials synthesized under high pressure such as H3S,1 LaH10,2 and the C–S–H system3 have opened possible routes to high-temperature superconductivity. The uranium–hydrogen binary system is among the potential candidates for such materials.4 Kruglov et al.4 synthesizied the predicted UH7 and UH8 in laser-heating diamond anvil cell (DAC) experiments, using UH3 as the precursor material. UH3 is a stable composition under ambient conditions5–8 and has important roles in nuclear technology and as a chemical hydrogen storage material.9–13 Therefore, exploring the high-pressure properties of UH3 is important for both fundamental research and industrial applications.5
At present, there is a fairly comprehensive understanding of UH3 under ambient condition. UH3 crystallizes in two structures: α-UH3 and β-UH3.14 The α-UH3 structure is metastable and can only be found at low temperatures.15 It transforms into stable β-UH3 at temperatures between 373 and 523 K.16 Taylor et al.15 simulated the phase transformation paths from α-UH3 to β-UH3. Tkach et al.17 stabilized α-UH3 with Zr and conducted experiments to study its electronic properties. β-UH3 transforms from a ferromagnetic (FM) state18 to a paramagnetic (PM) state at a Curie temperature of about 180 K.19 Zhang et al.16 calculated the electronic, mechanical, and thermodynamic properties of both UH3 phases under ambient conditions.
Zhang et al.20 calculated the mechanical and thermodynamic properties of α-UH3 under high pressures. According to DAC experiments,4,21α-UH3 transforms to β-UH3 when the pressure is increased to 5 GPa, and the x-ray diffraction signature of β-UH3 is preserved up 69 GPa.4 According to theoretical calculations by Taylor,22 ferromagnetism in β-UH3 vanishes when the unit-cell volume becomes about 50% of the equilibrium volume. In particular, the magnetization exhibits a sharp decrease from about 2.8 to 2.0 μB/U as the volume of the system increases from 32 to 36 Å3.22 These characteristics of the magnetization appear to be analogous to the isostructural transitions in 3d materials such as FeCO323 and LiFePO4.24 In Refs. 23 and 24, the structure was retained, while the spin state was transformed from a high-spin (HS, S = 2) to a low-spin (LS, S = 0) state under pressure, accompanied by volume collapse at the transition point. Actually, the magnetic order of β-UH3 remains ferromagnetic, and the impact of the transition on the volume is not known at present. Besides, β-UH3 contains 5f electrons,25,26 and this gives rise to exotic physics.27 Thus, it is intriguing to study the electronic structures of β-UH3 under high pressure.
In this work, we conduct first-principles calculations of the electronic structure of β-UH3 under high pressure. A local screened Coulomb correction (LSCC) approach28 is employed to evaluate the pressure-dependent local Coulomb interaction parameters in a self-consistent way. We predict an electronic structure transition of β-UH3 under high pressure and illustrate it from four perspectives. First, we find that the magnetization has a discontinuity around 32 Å3, suggesting a possible electronic structure transition. Second, we find that the calculated band structure of β-UH3 is reconstructed after 18 GPa. Third, after the transition, the density of states (DOS) becomes more dispersed. Fourth, we present an order parameter to quantify the electronic structure transition by calculating the 5f electron energy of the occupied states under pressure. Finally, we analyze the charge density difference to reveal the enhanced metallicity and calculate the volume drop after the transition.
II. COMPUTATIONAL METHODS
The space group of the stable β-UH3 structure is Pm
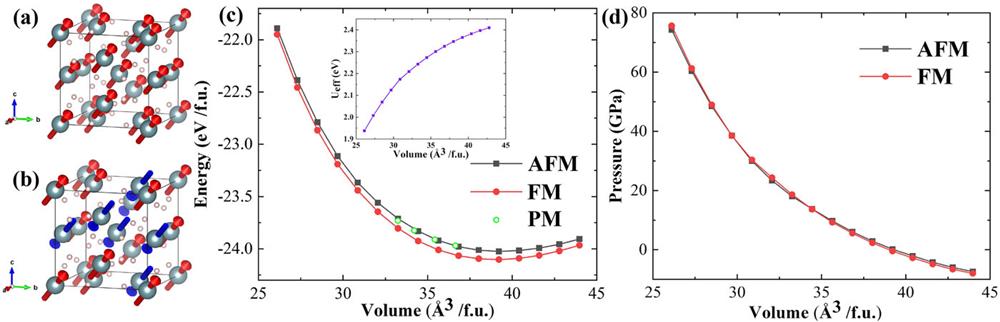
In the present ab initio calculations, we employ the Vienna Ab initio Simulation Package (VASP) based on density functional theory (DFT).32,33 The exchange-correlation functional is treated using the generalized gradient approximation (GGA) of Perdew, Burke, and Ernzerhof.34 The calculations use the projector-augmented wave (PAW)35 approach to describe the core electrons and their effects on valence orbitals. Spin–orbit coupling (SOC) is taken into account, with the direction of magnetic axis along (111). Ferromagnetic (FM) ordering is assumed in the simulation [Fig. 1(a)].
![]()
Figure 1.(a) and (b) Magnetic structures of the FM and AFM states, respectively, with the red and blue arrows denoting the magnetic axes. (c) Energy–volume relations of the FM, AFM, and PM states. The inset shows
DFT + U36 calculations are performed using the formulation of Dudarev et al.37 to account for the on-site Coulomb repulsion among the localized uranium 5f electrons. The total energy is of the form
To estimate the Curie temperature TC, we use the classical Heisenberg model in mean-field theory (MFA).38 The Curie temperature is given by
We recheck our electronic structure calculations using the orbital polarization (OP) limit of GGA + U.16
We set the plane-wave kinetic-energy cutoff to 650 eV, and the Brillouin zone is sampled with a special k-mesh generated by the Monkhorst–Pack scheme with a k-point spacing of 2π × 0.03 Å−1. The convergence tolerance is 10−6 eV for total energy, and all forces are converged to be less than 0.003 eV/Å.
III. RESULTS AND DISCUSSION
High-pressure experimental4,21 and theoretical5 studies suggest that there is no structural transition in β-UH3 up to 69 GPa. In our calculation, the structure remains Pm
We present the electron magnetization per uranium atom in Fig. 2. The discontinuity in magnetization is around 20 GPa, and the magnitude is about 0.14 μB/U. The pressure of 20 GPa corresponds to about 33 Å3. The reduction in magnetization indicates a possible electronic structure transition, and the abrupt change around 33 Å3 suggests an immediate elevation of metallicity.
![]()
Figure 2.Electron magnetization per uranium atom as a function of pressure for
To illustrate the transition, we calculate the band structures of β-UH3 at pressures up to 75 GPa, as plotted in Fig. 3. The band structures below 18 GPa have analogous characteristics, such as a “bandgap” along the Brillouin path X–R. We can also observe a gradual variation in the band structure from 24 to 75 GPa. Here, we characterize two types of electronic structure. One, E1, exhibits a “bandgap” along X–R, as exemplified by the band structures at pressures below 18 GPa [Figs. 3(a)–3(c)]. The other, E2, lacks this “bandgap,” as exemplified by the band structures at pressures above 24 GPa [Figs. 3(d)–3(f)]. From a comparison of E1 at 18 GPa [Fig. 3(c)] with E2 at 24 GPa [Fig. 3(d)], it can be seen that the “bandgap” along X–R undergoes a sudden collapse. The band structure is reconstructed along other Brillouin paths as well. This suggests the existence of a transition in electronic structure between 18 and 24 GPa.
![]()
Figure 3.Band structures of
We then calculate the DOS of β-UH3 under different pressures [Fig. 4(a)]. At each pressure, the DOS is relative to the Fermi energy. As can be seen from Fig. 4(a), the distribution below 18 GPa is relatively localized, while that above 24 GPa becomes more dispersed. In Figs. 4(b) and 4(c), the f electrons make the greatest contribution in both the E1 and E2 states around the Fermi energy. Hence, the dispersed behavior above 24 GPa, such as the emerging peaks around −1.5 eV at 24 GPa [Fig. 4(a)], can be attributed to the redistribution of 5f electrons.
![]()
Figure 4.(a) DOS of
To quantify the electronic structure transition, we propose the following order parameter:
![]()
Figure 5.5
We calculate the enthalpy of the distinct electronic states and determine the transition point at 21 GPa. At this pressure, we analyze the charge density difference between E1 and E2 states. As illustrated in Fig. 6, we select the (200) plane, the isosurface value is ±0.001 e/bohr,3 and blue and red colors represent areas losing and gaining electrons, respectively. The areas losing electrons are localized around uranium and hydrogen atoms, while the areas gaining most electrons are connected via the H–U(II)–U(II)–H channel. Hence, the E2 state is more itinerant than the E1 state; in other words, the metallicity is elevated after the electronic structure transition.
![]()
Figure 6.Charge density difference of
We recheck the electronic structure calculations with the OP limit of GGA + U.16 The ground state retains FM order within 70 GPa, and we can observe an electronic structure transition around 13 GPa (33 Å3) as well. This confirms the validity of the electronic structure transition under high pressure for β-UH3. Besides, the volume drops about 0.7% at the transition point, which is significantly less than the 10% drop for FeCO323 and the 3% drop for LiFePO4.24 This may explain why volume collapse has not reported in previous high-pressure experiments.4,21
To further verify the electronic structure transition in β-UH3, we suggest that detailed partial fluorescence yield x-ray absorption spectroscopy (PFY XAS) or resonant inelastic x-ray scattering (RIXS) spectra under pressure be performed in the future.
IV. CONCLUSIONS
In summary, using first-principles calculations, we have predicted an electronic structure transition in β-UH3 at about 20 GPa, and we have illustrated this from the perspectives of magnetization, band structure, DOS, and 5f electron energy. On the basis of the 5f electron energy, we propose an order parameter as a descriptor for the electronic structure transition. After the electronic structure transition, the itinerancy of the 5f electrons exhibits a sudden enhancement, accompanied by an abrupt elevation in metallicity. β-UH3 remains ferromagnetic after the electronic structure transition, and this transition has only a slight impact on the structure volume. Our calculations provide further understanding of the electronic properties of β-UH3 under high pressure.
ACKNOWLEDGMENTS
Acknowledgment. We acknowledge support from the National Key Research and Development Program of China under Grant No. 2021YFB3501503 and from the National Natural Science Foundation of China under Grant Nos. 12004048 and U1930401.
References
[1] A. P.Drozdov, M. I.Eremets, V.Ksenofontov, S. I.Shylin, I. A.Troyan. Conventional superconductivity at 203 kelvin at high pressures in the sulfur hydride system. Nature, 525, 73-76(2015).
[2] M.Ahart, M.Baldini, Z. M.Geballe, R. J.Hemley, Y.Meng, A. K.Mishra, M.Somayazulu, V. V.Struzhkin. Evidence for superconductivity above 260 K in lanthanum superhydride at megabar pressures. Phys. Rev. Lett., 122, 027001(2019).
[3] N.Dasenbrock-Gammon, M.Debessai, R. P.Dias, K. V.Lawler, R.McBride, A.Salamat, E.Snider, K.Vencatasamy, H.Vindana. Room-temperature superconductivity in a carbonaceous sulfur hydride. Nature, 586, 373-377(2020).
[4] A. F.Goncharov, E.Greenberg, N.Holtgrewe, S.Jiang, I. A.Kruglov, A. G.Kvashnin, S. S.Lobanov, A. R.Oganov, V. B.Prakapenka, A. V.Yanilkin. Uranium polyhydrides at moderate pressures: Prediction, synthesis, and expected superconductivity. Sci. Adv., 4, eaat9776(2018).
[5] M.Li, X.Wang, P.Zhang, F.Zheng. Crystal structure prediction of uranium hydrides at high pressure: A new hydrogen-rich phase. Phys. Lett. A, 382, 2959-2964(2018).
[6] B.Bai, X.Chen, T.Fa, D.Li, M.Liu, M.Liu, W.Mo, Y.Shi, X.Wang. First-principles comprehensive study of electronic and mechanical properties of novel uranium hydrides at different pressures. Prog. Nat. Sci.: Mater. Int., 30, 251-259(2020).
[7] L.Andrews, G. P.Kushto, M.Neurock, P. F.Souter. Experimental and theoretical evidence for the formation of several uranium hydride molecules. J. Am. Chem. Soc., 119, 1682-1687(1997).
[8] L.Andrews, L.Gagliardi, R. H.Lindh, J.Raab, X.Wang. A combined experimental and theoretical study of uranium polyhydrides with new evidence for the large complex UH4(H2)6. J. Phys. Chem., 111, 6383-6387(2007).
[9] P.Camp, M. H.Chang, S.Cho, D.Chung, H.Chung, K. J.Jung, H.-G.Kang, C. S.Kim, K. H.Kim, D.Koo, H.Lee, J.Lee, S.Paek, H.Yoshida, S.-H.Yun. Hydriding and dehydriding characteristics of small-scale DU and ZrCo beds. Fusion Eng. Des., 88, 2276-2279(2013).
[10] A.Banos, N. J.Harker, T. B.Scott. A review of uranium corrosion by hydrogen and the formation of uranium hydride. Corros. Sci., 136, 129-147(2018).
[11] H.Ju, W.Kim, H.Yoo. A numerical comparison of hydrogen absorption behaviors of uranium and zirconium cobalt-based metal hydride beds. Solid State Ionics, 262, 241-247(2014).
[12] F. D.Manchester, A.San-Martin. The H-U (hydrogen-uranium) system. J. Phase Equilib., 16, 263-275(1995).
[13] J.Bloch. The hydriding kinetics of activated uranium powder under low (near equilibrium) hydrogen pressure. J. Alloys Compd., 361, 130-137(2003).
[14] F. H.Ellinger, R. N. R.Mulford, W. H.Zachariasen. A new form of uranium hydride. J. Am. Chem. Soc., 76, 297(1954).
[15] R. S.Lillard, T.Lookman, C. D.Taylor. Ab initio calculations of the uranium–hydrogen system: Thermodynamics, hydrogen saturation of
[16] Y.Lu, B.Wang, Y.Yang, P.Zhang, Y.Zhang. Electronic, mechanical and thermodynamic properties of
[17] A. V.Andreev, M.Cieslar, M.Divi?, D.Drozdenko, L.Havela, N.-T. H.Kim-Ngan, D.Kriegner, Z.Matěj, M.Paukov, I.Tkach, I.Turek, B.Vondrá?ková. Electronic properties of
[18] W.Suski, R.Troc. The discovery of the ferromagnetism in U(H, D)3: 40 years later. J. Alloys Compd., 219, 1(1995).
[19] V.Buturlim, M.Divis, M.Dopita, L.Havela, L.Kyvala, D.Legut, J.Prchal, I.Turek, J.Valenta. Pressure variations of the 5
[20] H.Jiang, H.-L.Shi, Y.-H.Su, C.Zhang, G.-H.Zhong. Mechanical and thermodynamic properties of
[21] M.Brill, I.Halevy, S.Salhov, I.Yaar, S.Zalkind. High pressure study of
[22] C. D.Taylor. Characterizing electronic structure motifs in
[23] R.Ahuja, B.Johansson, W.Luo, H.Shi. First-principles calculations of the electronic structure and pressure-induced magnetic transition in siderite FeCO3. Phys. Rev. B, 78, 155119(2008).
[24] E.Bykova, I.Efthimiopoulos, M.Koch-Müller, C.McCammon, J.Müller, M.Nú?ez Valdez, M.Taran, M.Wilke. Evidence for a pressure-induced spin transition in olivine-type LiFePO4 triphylite. Phys. Rev. B, 97, 184405(2018).
[25] E.Colineau, R.Eloirdi, T.Gouder, F.Huber, D.Kolberg, J.Rebizant, F.Wastin. Electronic structure of UH3 thin films prepared by sputter deposition. Phys. Rev. B, 70, 235108(2004).
[26] G.Zwicknagl. 5
[27] L.Huang, H.Lu. Pressure-driven 5
[28] H.Jiang, Y.-C.Wang. Local screened Coulomb correction approach to strongly correlated
[29] R. E.Rundle. The structure of uranium hydride and deuteride. J. Am. Chem. Soc., 69, 1719-1723(1947).
[30] R. E.Rundle. The hydrogen positions in uranium hydride by neutron diffraction. J. Am. Chem. Soc., 73, 4172-4174(1951).
[31] J.Grunzweig-Genossar, M.Kuznietz, B.Meerovici. Nuclear magnetic resonance in uranium hydride and deuteride. Phys. Rev. B, 1, 1958(1970).
[32] J.Furthmüller, G.Kresse. Efficient iterative schemes for
[33] H. J.Monkhorst, J. D.Pack. Special points for Brillouin zone integrations. Phys. Rev. B, 13, 5188(1976).
[34] K.Burke, M.Ernzerhof, J. P.Perdew. Generalized gradient approximation made simple. Phys. Rev. Lett., 77, 3865(1996).
[35] P. E.Bl?chl. Projector augmented-wave method. Phys. Rev. B, 50, 17953(1994).
[36] O. K.Andersen, V. I.Anisimov, J.Zaanen. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B, 44, 943(1991).
[37] G. A.Botton, S. L.Dudarev, C. J.Humphreys, S. Y.Savrasov, A. P.Sutton. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B, 57, 1505(1998).
[38] A. H.Morrish. The Physical Principles of Magnetism(1965).
[39] X.Gao, D.-Y.Lin, H.-F.Song, F.Tian, Y.-F.Zhao. A structural modeling approach to solid solutions based on the similar atomic environment. J. Chem. Phys., 153, 034101(2020).
[40] B.Dorado, P.Garcia. First-principles DFT+U modeling of actinide-based alloys: Application to paramagnetic phases of UO2 and (U, Pu) mixed oxides. Phys. Rev. B, 87, 195139(2013).


Set citation alerts for the article
Please enter your email address

© Copyright 2018-2021 | Chinese Laser Press. All Rights Reserved 沪ICP备15018463号-20