
Xing Li, Xiaohua Zhang, Yong Liu, Guochun Yang. Bonding-unsaturation-dependent superconductivity in P-rich sulfides[J]. Matter and Radiation at Extremes, 2022, 7(4): 048402
- Matter and Radiation at Extremes
- Vol. 7, Issue 4, 048402 (2022)
Abstract
I. INTRODUCTION
Pressure, as an external control method, can stabilize exotic compounds that defy the traditional understandings of stoichiometry, structural form, and properties in ambient conditions.1–4 One of the most representative cases of this phenomenon is the burgeoning field of high-pressure superconducting hydrides, which have remarkably high critical temperatures (Tc > 200 K).5–9 Within these compounds, H atoms or H and other atoms can form rigid covalent motifs, exhibiting diverse coordination configurations such as symmetric S–H–S bonds (twofold coordination) in H3S, 1D nanotubes via threefold coordination in HfH9,10 2D penta-graphene containing twofold and threefold coordination in HfH10,11 and 3D cages with fourfold coordination in LaH10 and CaH6.12–14 These coordination patterns are much beyond the bonding limit of single-electron H atoms at ambient pressure, and their corresponding covalent bonds thus possess fewer electrons than a conventional shared electron pair, appearing to be unsaturated. Strikingly, these kinds of H frameworks have been demonstrated to be able to induce strong electron–phonon coupling (EPC).15 Thus far, the understanding of unsaturated-bonding-related superconductivity is quite limited. However, bonding unsaturation could provide a new approach to the design of superconductors.
Phosphorus (P) and sulfur (S), as adjacent elements in the Periodic Table, have moderate electronegativity as well as a strong ability to form rich P/S allotropes16–19 and P–S compounds.20–24 Interestingly, the majority of P–S compounds exhibit cage-like structures,21–24 and this bestows them with intriguing properties and gives them a broad range of potential applications.25,26 For instance, P4S3, which has strong resistance to volume changes during electrochemical reactions, is considered as an ideal anode material with high capacity and cycling stability for Li+/Na+/K+ storage, providing a reversible capacity of up to 1266 mAh g−1 in Li–ion batteries.25 P4S10, having high chemical and electrochemical stability, can act as a protective layer for anodes, overcoming the corrosion and dendrite problems of Li metal anodes.26
In ambient conditions, P and S, which have 3s23p3 and 3s23p4 valence electrons, usually show three-/two-coordination in their covalent compounds, satisfying the well-known octet rules. However, in their high-pressure allotropes and compounds, their coordination numbers can be boosted to 8 and 6, respectively,17–19,27 resulting in the formation of diverse covalent frameworks with notable bonding unsaturation, contrasting with the classical covalent bonds formed by sharing an electron pair. This unique bonding means that P and S are two significant candidate elements for high-Tc superconductors.28–31
The first experimental validation of a superconducting hydride (Tc > 200 K) was conducted with H3S, which has a record Tc of 203 K at 155 GPa,32 and this consists of nested SH6 octahedral networks with each other.4 Moreover, monoclinic (C2/m) PH3, with a P–H covalent framework and six-coordinated P, exhibits a maximum Tc of 83 K at 200 GPa in P-H compounds.30 Based on the difference in valence electrons, the S atoms in H3S can be partially substituted with non-metallic atoms from the IV-, V-, VI-, or VII-A groups,33–36 and this can be used to modulate the position of the van Hoff singularity with respect to the Fermi level (EF), raising the Tc value. In fact, the bonding unsaturation in doped H3S can also be tuned. Interestingly, the Tc value has been found to increase significantly to 290 K at 250 GPa after replacing 7.5% of the S atoms with P atoms.34 To date, numerous binary and ternary P- and S-based compounds have been predicted to exhibit excellent superconductivity associated with their various covalent networks.37–41
Pressure-induced P–S compounds have also attracted great research interest due to the unique bonding behavior of P and S atoms. In particular, two pioneering works explored the exotic properties of P–S compounds at high pressure.42,43 Li et al. found that as the pressure increases, the stable phases (e.g., P2S7 and P4Sx, x = 3–10) at 0 GPa gradually decompose, and above 8 GPa, the only predicted stable phase is R-3m PS2; this was confirmed by experiments.42 More interestingly, R-3m PS2, which is isostructural to 3R MoS2, is a van der Waals layered compound with a predicted Tc of ∼11 K at 0 GPa. In addition, Liu et al. found several P–S compounds including P3S, P2S, PS, PS2, and PS3, showing abundant S- and P-centered polyhedrons (e.g., SP6, SP4, SP3, PS6, and PS3).43 However, there is a difference regarding the stability of high-pressure PS2 phases: C2/m and P-3m1 PS2 were found to stable from 5 to 200 GPa in Liu et al.’s work,43 but the synthesized R-3m phase disappears.42 In summary, the veil over the nature and behavior of P–S compounds at high pressure has not yet been fully lifted.
Taking into account the peculiar bonding behavior of P and S atoms and the imperfect high-pressure P–S phase diagram, herein, we perform a comprehensive structural search with various stoichiometries PxSy (x/x + y = 0–1, 38 compositions) to explore as-yet untapped P–S compounds and their electronic properties. Three unknown P-rich phases, Cmcm P5S, P-31m P8S, and P-1 P11S, were identified, in which both P and S atoms present sixfold octahedral coordination with unsaturated covalent bonds, inducing superconductivity. Furthermore, in P-rich P–S compounds and several MH9 hydrides, a positive correlation was established between unsaturated bonding and the superconducting transition temperature, indicating the feasibility of modulating bonding saturation to improve superconductivity. In addition to the three proposed PS2 phases, a new low-pressure P-3m1 PS2 phase was found, representing a missing link in the phase-transition sequence with increasing pressure.
II. COMPUTATIONAL DETAILS
An intelligence-based particle swarm optimization algorithm using the CALYPSO software package has been demonstrated to be an important way to discover new structures.7 In this work, structures with various PxSy compositions (x = 1, y = 1/12, 1/11-1/3, 3/8, 2/5, 1/2, 3/5, 2/3, 3/4, 4/5, 1, 5/4, 4/3, 3/2, 5/3, 7/4, 2, 9/4, 5/2, 8/3, 3, 7/2, 4, 5-12) were extensively searched with simulation cell sizes of 1–4 formula units at 25, 50, 100, and 200 GPa. Structural relaxation and electronic structure calculations were carried out within framework of density functional theory with the Perdew–Burke–Ernzerhof generalized gradient approximation,44 as implemented in the Vienna Ab initio Simulation Package.45 Projector augmented waves with 3s23p3 and 3s23p4 electrons were used as valences for P and S atoms, respectively.46 The reliability of our adopted pseudopotentials was confirmed by the perfect fitting of the Birch–Murnaghan equation of states with the full-potential linearized augmented-plane-wave method [Fig. S3(a)]. A plane-wave cutoff energy of 600 eV and a Monkhorst–Pack scheme with a k-point grid of 2π × 0.03 Å−1 in the Brillouin zone were used to ensure an enthalpy convergence of less than 1 meV/atom. The electron localization function (ELF) is used to describe and visualize the chemical bonds of the stable compounds.47 The integrated crystal orbital Hamilton population (ICOHP) was calculated using the LOBSTER software package.48 Phonon-dispersion curves were calculated using the supercell finite-displacement method in the Phonopy software package.49
The EPC calculations were carried out within the framework of density-functional perturbation theory, as executed in the Quantum ESPRESSO package.50 We employed ultrasoft pseudopotentials, with 3s23p3 and 3s23p4 considered as the valence electrons for P and S, respectively. A kinetic-energy cutoff of 70 Ry and Gaussians of width 0.02 Ry were applied. To reliably calculate the EPC in metallic systems, we need to sample dense k-meshes for the electronic Brillouin zone integration and enough q-meshes to evaluate the average contributions from the phonon modes. Dependent on specific structures of the stable compounds, different sizes of k- and q-meshes were used: 15 × 15 × 9 k-meshes and 5 × 5 × 3 q-meshes for P2S in the C2/m-I structure, 12 × 12 × 3 k-meshes and 3 × 3 × 3 q-meshes for P3S in the C2/m structure, 15 × 15 × 6 k-meshes and 5 × 5 × 2 q-meshes for P5S in the Cmcm structure, 8 × 8 × 12 k-meshes and 4 × 4 × 6 q-meshes for P8S in the P-31m structure, and 12 × 12 × 9 k-meshes and 4 × 4 × 3 q-meshes for P11S in the P-1 structure.
III. RESULTS AND DISCUSSION
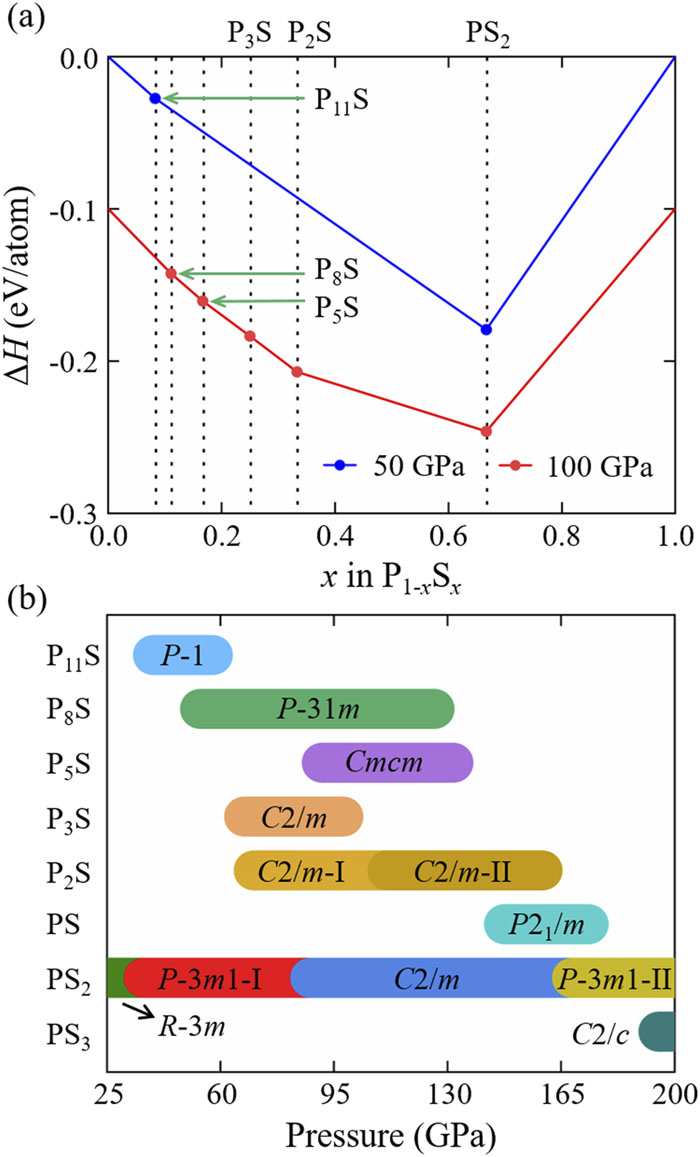
To explore the phase diagram of the P–S system, an extensive structure search was performed by focusing on the 38 stoichiometries. The relative stabilities of the PxSy compounds were determined using their formation enthalpies with respect to the solid phases of P27 and S18,51 at the considered pressures (Fig. S1). We not only reproduced the theoretically proposed42,43C2/m P3S, C2/m-I P2S, C2/m-II P2S, P21/m PS, R-3m PS2, C2/m PS2, P-3m1 PS2, and C2/c PS3 structures at high pressures, but also identified three hitherto unknown P-rich compounds: Cmcm P5S, P-31m P8S, and P-1 P11S [Fig. 1(a)], as well as a new P-3m1 PS2 phase (labeled as P-3m1-I, Fig. S2).
![]()
Figure 1.(a) Convex hull constructed with thermodynamically stable P–S compounds at 50 and 100 GPa. For clarity, we have offset the formation enthalpy by −0.1 eV for 100 GPa. The full convex hull is shown in Fig. S1. (b) Pressure–composition phase diagram of stable P–S compounds.
The optimized lattice parameters of R-3m PS2 (a = 3.165 Å and c = 15.572 Å) were found to be in good agreement with the experimentally obtained values (a = 3.149 Å, and c = 15.501 Å) at 14 GPa;42 additionally, the use of different exchange–correlation functionals did not affect the thermodynamic stabilities of the phases proposed here [Fig. S3(b)], confirming the reliability of our calculations. Both C2/m and C2/c PS3 were predicted by Liu et al. to be thermodynamically stable.43 However, in our work, we found that only the C2/c phase is thermodynamically stable. In other words, C2/m PS3 is unstable against decomposition into S and PS2 in our considered pressure range of 25–200 GPa, as shown in Fig. S2(b). All the predicted structures are dynamically stable due to the absence of any imaginary frequencies in the phonon dispersion curves (Fig. S4). The stabilizing pressure ranges of the identified phases are illustrated in Fig. 1(b), and this provides useful information for experiments.
There are obvious differences in the stable stoichiometries and structural characters between high and ambient pressures. Most of the P–S compounds are S-rich stoichiometries with the structural features of a cage-like molecular crystal in P4Sx (x = 5, 7, 9, and 10) and infinite wrinkled chains in P2S7 at ambient pressure.20–24 Increasing the S content in the compounds induces a gradual transformation of coordination configuration from triangular cone to tetrahedron for the P atoms, and this is accompanied by an increase of the coordination number (Fig. S5). In sharp contrast, at high pressure, P-rich stoichiometries are stabilized (PxS, x = 5, 8, and 11). Notably, these P-rich compounds exhibit similar P–S frameworks, including the reported P2S and P3S, which can be viewed as derivatives of S-doped simple cubic (sc) P [Figs. 2(a)–2(c)]. In other words, both the P and S atoms in these structures maintain a six-coordinated octahedron configuration independent of the P/S ratio, but the S-centered octahedra demonstrate different coordination atoms, such as four P atoms and two S atoms in P2S, or mixed four P and two S atoms as well as six P atoms in P3S, as well as six P atoms in PxS (x = 5, 8, and 11) at 100 GPa (Fig. S6).
![]()
Figure 2.Crystal structures of (a)
The P/S ratio can be used to effectively modulate the orientation and distribution of S-centered octahedra. Specifically, in P2S, P3S, and P11S, S-centered octahedra are distributed horizontally with S atoms in the same layer, leading to the formation of alternating P layers perpendicular to the horizontal plane [Figs. S6(a), S6(b), and S6(f)]. In P5S and P8S, the central S atoms are alternately distributed in different directions, inhibiting the formation of the P layer [Figs. S6(c)–S6(e)]. As a result, different connections appear between the S-centered octahedra, including mixed edge/vertex sharing in P2S and P3S, vertex sharing in P5S, and P2 pair sharing in P8S and P11S. In the PxS (x = 5, 8, and 11) compounds, the P–P and P–S bond lengths are in the range 2.12–2.17 Å at 100 GPa, which is close to the P–P (2.20 Å) and P–S (2.09 Å) bond lengths in black phosphorus and P4S3 at ambient pressure,52,53 indicating that covalent P–P/S bonds are responsible for the stabilization, as demonstrated by the ELF [Fig. 4(a) and Fig. S7].
As noted in the introduction to this paper, the previously reported stable phases of PS2 are discrepant under pressure. Aside from the R-3m phase being stable in the range 8–20 GPa,42 the C2/m and P-3m1 phases are found to be stable in the ranges 5–178 and 178–200 GPa,43 respectively. Here, we not only reproduce the stable R-3m, C2/m, and P-3m1 phases, but we also identify a new P-3m1-I phase, which stabilizes from 36.3 to 87.6 GPa; this is an intermediate phase between the R-3m and C2/m phases. Notably, the discovery of the P-3m1-I phase settles the contradiction regarding the PS2 phase diagram and provides a plausible description of the phase-transition process of PS2 with pressure.
For R-3m PS2 with an ABC stacking order, pressure makes the sublayers slide along ab plane into AA stacking, accompanying the transition from the R-3m phase to the P-3m1-I phase (Fig. S8). With further compression, the sublayers in the P-3m1-I phase move away from each other along the a axis until transitioning into C2/m symmetry with staggered sublayers in the vertical direction [Figs. 2(d) and 2(e)]. Then, the PS2 sublayers in the C2/m phase move toward each other along the a axis with increasing pressure. Finally, AA stacking P-3m1-II PS2 is obtained above 168.3 GPa [Figs. 2(e) and 2(f)], in which both S and P atoms form a covalent octahedron coordination (e.g., S-centered P3S3 and PS6). Overall, the lattice parameters and distances between S atoms of adjacent sublayers progressively decrease with pressure (Fig. S9). Meanwhile, the charge is redistributed and the interlayered S–S interaction is enhanced, as supported by the ELF (Fig. S10), resulting in a transformation of PS2 from a van der Waals layered structure to a 3D covalent framework.
One prominent structural characteristic is the formation of covalent octahedron coordination for P and S atoms in P-rich P–S compounds at high pressure. This is mainly attributed to the head-to-head overlap of three p-orbital wave functions [Fig. 3(a)], as verified below. Here, we follow the principle from simplicity to complexity. First, sc P is selected due to the identical orthometric basis vector between the crystal structure and the covalent bond orientation. The calculated ICOHP exhibits dominant bonding interactions between the px and px, py and py, and pz and pz orbitals along the three bonding directions (Fig. S11). Meanwhile, P 3s electrons are involved in bonding with the 3p electrons of corresponding orientation. A similar bonding pattern is also demonstrated in Im-3m H3S (Fig. S12). Similarly, PxS compounds follow the bonding pattern of P8S (Figs. S13 and S14) by excluding the basis-vector difference between the atomic p orbitals and the covalent bond orientations.
![]()
Figure 3.(a) Schematic diagram of the bonding mechanism in sc P. (b) Average bonding unsaturation, (c)
The corresponding covalent bonds for P atoms in these P-rich compounds have unsaturated bonding due to there being five valence electrons from each P atom for six bonds. Here, we propose a standard for evaluating the unsaturation of covalent bonds in these compounds. Each S or P atom has 6 or 5 valence electrons, respectively. On average, a single S atom can provide one electron per covalent bond in a six-coordinated octahedron, whereas a P atom can provide 5/6 of an electron. Therefore, the number of electrons is (1 + 1)e− in each S–S bond, (1 + 5/6)e− in each P–S bond, and (5/6 + 5/6)e− in each P–P bond; the S–S bonds are saturated (e.g., a two-centered two-electron bond), and the other two are unsaturated.
For ease of measuring the covalent unsaturation of these three kinds of bonds, the saturation of each S–S bond is normalized to 1—meaning that the saturations of P–S and P–P bonds are 0.917 and 0.833, respectively—and the bonding unsaturation is defined to be the difference between 1 and the saturation, e.g., 0, 0.083, and 0.167 for S–S, P–S, and P–P bonds, respectively. Correspondingly, the average bonding unsaturation of these five P-rich P–S compounds is 0.106 for P2S, 0.125 for P3S, 0.139 for P5S, 0.148 for P8S, and 0.153 for P11S [Fig. 3(b)], the detailed calculations can be found in
The calculated projected densities of states (PDOS) illustrate that P5S, P8S, and P11S are metallic on account of the large electronic contributions at EF [Fig. 4(b) and Fig. S15]. The considerable orbital overlap between P and S atoms (3p–3p, and 3s–3s) also supports their strong covalent interaction. With respect to P/S 3s states, the 3p states have a much larger DOS at EF, dominating the metallicity of these P–S compounds. Notably, their DOS amplitude at EF (
![]()
Figure 4.(a) ELF, (b) PDOS, (c) projected electronic band structure, (d) PHDOS and Eliashberg spectral function and
On account of the identical coordination modes and distinct bonding unsaturation values caused by different P/S ratios in P-rich compounds (e.g., P2S, P3S, P5S, P8S, and P11S), they provide an ideal model for the study of bonding-unsaturation-related superconductivity. To accurately describe this relationship, we calculated their Tc values at an identical pressure (e.g., 100 GPa) by employing the Allen–Dynes-modified McMillan equation.54 Here, the Tc values of the previously proposed C2/m-I P2S and C2/m P3S structures are recalculated for better comparability. The resulting Tc values (2.63 K for C2/m-I P2S, 3.06 K for C2/m P3S) are in accordance with the reported values (Fig. S17).43 Interestingly, the value of Tc increases monotonously with the increasing average bonding unsaturation from P2S to P11S, indicating an evident positive correlation between bonding unsaturation and Tc values [Fig. 3(c)]. Furthermore, their λ values present the same evolution trend with Tc values; the trend in ωlog is opposite, manifesting a decisive effect of λ on the variation of Tc value.
The increasing λ value can be principally ascribed to the elevated
To further examine the effect of bonding unsaturation on superconductivity, we inspected reported superconducting TM hydrides based on these criteria: they must have the same H cage, different electrons donated by TM atoms that affect bonding saturation, and pressure differences within 30 GPa to minimize the effects of pressure. Among the three well-known TMHx clathrates (LaH10-type with H32 cage, YH9-type with H29 cage, and CaH6-type with H24 cage),57 only YH9-type clathrates can meet these requirements. Here, we assume that the smaller the number of TM valence electrons, the higher the average bonding unsaturation of the H cage. This is also confirmed by the calculated charge-transfer amount (Fig. S16).
As shown in Fig. 3(f) and Fig. S16, with increasing average bonding unsaturation, the Tc values of TMH9 (M = Y, Th, and Pr) present an increasing trend.57–59 This is also similar to the Tc evolution of H3S after incorporating a small number of Si/C/P atoms, i.e., doped H3S with more electron-deficient atoms than the S atom can elevate the bonding unsaturation and the corresponding Tc values (e.g., Tc = 283 K at 230 GPa, 220 K at 225 GPa, and 212 K at 200 GPa for 4% Si, 7% C, and 6.25% P-doped H3S with average bonding unsaturations of 0.257, 0.262, and 0.255, respectively, higher than H3S).33,60,61 These results provide further support for the relationship between bonding unsaturation and superconductivity. It is noteworthy that our work opens the door to greater understanding of bonding-unsaturation-dependent superconductivity; further efforts are needed for a deeper understanding.
To explore the origin of superconductivity in these materials, we take P8S as an example due to it having the highest structural symmetry and the widest stabilizing pressure range among the P-rich compounds. The projected electronic band structure of P8S reveals obvious P–S hybridization across the whole Brillouin zone [Fig. 4(c)]. Moreover, several bands crossing EF demonstrates steep dispersion along the Γ–A, A–H, and L–H directions, as well as a flat feature around the Κ point [Fig. 4(c)]. The calculated EPC parameter (λ) of P8S is 0.47 at 100 GPa [Fig. 4(d)], and this is comparable to the values of 0.52 for C2/m S3PH at 150 GPa,37 0.59 for Cmcm PH3 at 100 GPa,31 and 0.42 for Pnma H4S3 at 140 GPa,62 and it is slightly lower than the value of 0.64 for Cmc21 SP2H at 150 GPa.37 Based on the Eliashberg spectral function α2F(ω) and the phonon density of states (PHDOS), we can establish that the phonons derived from P and S atoms show evident coupling across the whole phonon frequency range due to their similar mass and strong covalent interactions; the phonons in the frequency range 10.0–18.9 THz make the main contribution (59%) to the total λ, and these correspond to two large S- and P-dominated PHDOS peaks [Fig. 4(d)]. In particular, above 14.5 THz, there appears to be a rapid increase in λ, resulting in a large contribution of 40%; this is also illustrated in the phonon dispersion curves [Fig. 4(e)]. Such a strong EPC is thus mainly associated with P-atom vibrations and local phonon softening at the A point. Based on the Allen–Dynes-modified McMillan equation,54 the Tc value of P8S is estimated to be 4.15 K at 100 GPa with a typical Coulomb pseudopotential parameter of μ* = 0.1.63,64 Combining this with the electronic band structure, we can draw the conclusion that the superconductivity of P8S principally comes from the coupling between the P/S 3p electrons and the P-/S-derived phonons.
To obtain insight into the pressure dependence of the superconductivity of P8S, we investigated its superconductivity in the pressure range 55–125 GPa. The variation of Tc and the two key parameters (λ and ωlog) with pressure are displayed in Fig. 4(f). As the pressure decreases, Tc gradually increases, reaching a maximum of 6.0 K at 55 GPa. Below 100 GPa, the variation of λ is linearly related to Tc; this is in sharp contrast to ωlog, indicating that λ dominates the evolution of Tc. Above 100 GPa, the value of λ increases slightly, whereas ωlog decreases significantly, and this is responsible for the evolution of Tc. Overall, the pressure dependence of Tc is the result of competition between λ and ωlog.
IV. CONCLUSIONS
In summary, we performed an extensive first-principles structural search of P–S systems under high pressure and identified three stable P-rich compounds (Cmcm P5S, P-31m P8S, and P-1 P11S) and a new low-pressure PS2 phase with P-3m1 symmetry, which acts as an intermediate phase between the R-3m and C2/m phases, completing the understanding of the phase-transition process of PS2. All the P-rich PxS (x > 1) compounds have a common covalent framework, in which P and S atoms form an octahedral coordination via head-to-head hybridization of P/S 3p orbitals and unsaturated covalent bonds. More interestingly, the variation in bonding unsaturation induced by different P contents is nearly linearly related to the
SUPPLEMENTARY MATERIAL
ACKNOWLEDGMENTS
Acknowledgment. The authors acknowledge funding from the Natural Science Foundation of China (Grant Nos. 21873017 and 21573037), the Natural Science Foundation of Hebei Province (Grant Nos. A2019203507 and B2021203030), the Postdoctoral Science Foundation of China (Grant No. 2013M541283), and the Natural Science Foundation of Jilin Province (Grant No. 20190201231JC). This work was carried out at the National Supercomputer Center in Tianjin, and the calculations were performed on TianHe-1 (A).
References
[1] P.Gao, H.Gou, K.Hao, G.Liu, S.Liu, W.Lu, J.Lv, H.Wang, G.Yang, M.Zhou. Phys. Rev. Lett., 128, 106001(2022).
[2] T.Cui, D.Li, Y.Liu, R.Wang, Z.Wang. Nat. Commun., 13, 412(2022).
[3] X.Cai, J.Chen, Y.Chen, H.Du, X.Feng, H.Liu, S. A. T.Redfern, B.Sun, H.Wang, Y.Xie. Phys. Rev. B, 99, 184106(2019).
[4] T.Cui, D.Duan, X.Huang, D.Li, B.Liu, Y.Liu, F.Tian, W.Tian, H.Yu, Z.Zhao. Sci. Rep., 4, 6968(2014).
[5] R. J.Needs, A. R.Oganov, C. J.Pickard, Q.Zhu. Nat. Rev. Mater., 4, 331-348(2019).
[6] M. I.Eremets, I.Errea, C. J.Pickard. Annu. Rev. Condens. Matter Phys., 11, 57-76(2020).
[7] J.Lv, Y.Ma, Y.Wang, L.Zhang. Nat. Rev. Mater., 2, 17005(2017).
[8] M.Hanfland, A. G.Ivanova, I. A.Kruglov, A. G.Kvashnin, I. S.Lyubutin, K. S.Pervakov, A. V.Sadakov, D. V.Semenok, O. A.Sobolevskiy, I. A.Troyan. Mater. Today, 48, 18-28(2021).
[9] G.Yang, X.Zhang, Y.Zhao. Wiley Interdiscip. Rev.: Comput. Mol. Sci, 12, e1582(2022).
[10] S.Botti, J.Chen, W.Cui, K.Gao, J.Hao, Y.Li, C.Liu, M. A. L.Marques, J.Shi, Q.Wang. Phys. Rev. B, 104, 214511(2021).
[11] T.Cui, D.Duan, X.Feng, S.Jiang, V. Z.Kresin, C. J.Pickard, S. A. T.Redfern, H.Song, H.Xie, Y.Yao, Z.Zhang. Phys. Rev. Lett., 125, 217001(2020).
[12] N. W.Ashcroft, R. J.Hemley, R.Hoffmann, H.Liu, I. I.Naumov. Proc. Natl. Acad. Sci. U. S. A., 114, 6990(2017).
[13] M.Ahart, M.Baldini, Z. M.Geballe, R. J.Hemley, Y.Meng, A. K.Mishra, M.Somayazulu, V. V.Struzhkin. Phys. Rev. Lett., 122, 027001(2019).
[14] T.Iitaka, Y.Ma, K.Tanaka, J. S.Tse, H.Wang. Proc. Natl. Acad. Sci. U. S. A., 109, 6463(2012).
[15] A. S.Coolidge, H. M.James. J. Chem. Phys., 1, 825-835(1933).
[16] F.Bachhuber, R.Dronskowski, T.Nilges, A.Pfitzner, P.Schmidt, J.von Appen, R.Weihrich. Angew. Chem., Int. Ed., 53, 11629-11633(2014).
[17] Y.Akahama, H.Fujihisa, H.Fukui, N.Hirao, Y.Ohishi, Y.Ozawa, T.Sugimoto. Phys. Rev. B, 86, 024109(2012).
[18] R. G.Greene, H.Luo, A. L.Ruoff. Phys. Rev. Lett., 71, 2943(1993).
[19] C. W.Glass, A. R.Oganov. J. Chem. Phys., 124, 244704(2006).
[20] A.Pfitzner, T.R?dl, J.Senker, J.Wack, R.Weihrich. Angew. Chem., Int. Ed., 50, 10996-11000(2011).
[21] S. v.Houten, Y. C.Leung, A.Vos, J.Waser, E. H.Wiebenga, G. A.Wiegers. Acta Crystallogr., 10, 574-582(1957).
[22] P.Leibnitz, B.Wallis, G.-U.Wolf. Z. Anorg. Allg. Chem., 588, 139-146(1990).
[23] R.Botterweg, R.Olthof, F.van Bolhuis, A.Vos. Acta Crystallogr., 19, 864-867(1965).
[24] S.Van Houten, E. H.Wiebenga. Acta Crystallogr., 10, 156-160(1957).
[25] Z.Ju, H.Li, H.Li, D.Wei, Y.Wei, J.Yin, S.Zeng, W.Zhao. J. Energy Chem., 50, 187-194(2020).
[26] Y.Huang, Z.Jin, M.Li, Q.Li, X.Liu, A.Wang, W.Wang, Y.Yang. J. Energy Chem., 41, 27-33(2020).
[27] H.Katzke, P.Tolédano. Phys. Rev. B, 77, 024109(2008).
[28] P.Avery, T.Bi, Z.Falls, A.Shamp, T.Terpstra, E.Zurek. J. Am. Chem. Soc., 138, 1884-1892(2016).
[29] M.Amsler, L.Boeri, J. A.Flores-Livas, S.Goedecker, E.Gross, C.Heil, G.Profeta, A.Sanna, C.Wolverton. Phys. Rev. B, 93, 020508(2016).
[30] G.Gao, Y.Li, H.Liu, I. I.Naumov, J. S.Tse. J. Phys. Chem. C, 120, 3458-3461(2016).
[31] G.Fang, X.Li, Y.Li, G.Liu, C.Pei, L.Wang, K.Yang, Y.Yuan, H.Zheng. Natl. Sci. Rev., 6, 524-531(2019).
[32] A. P.Drozdov, M. I.Eremets, V.Ksenofontov, S. I.Shylin, I. A.Troyan. Nature, 525, 73-76(2015).
[33] R. P.Dias, Y.Ge, R. J.Hemley, Y.Yao, F.Zhang. Mater. Today Phys., 15, 100330(2020).
[34] Y.Ge, Y.Yao, F.Zhang. Phys. Rev. B, 93, 224513(2016).
[35] F.Fan, B. M.Klein, M. J.Mehl, D. A.Papaconstantopoulos. J. Phys. Chem. Solids, 99, 105-110(2016).
[36] M.Amsler. Phys. Rev. B, 99, 060102(2019).
[37] T.Bi, N.Geng, E.Zurek. J. Phys. Chem. C, 126, 7208(2022).
[38] T.Iitaka, B.Jiang, H.Li, X.Li, Y.Sun, Y.Tian, Y.Xie, X.Zhong. Phys. Rev. B, 101, 174102(2020).
[39] T.Cui, D.Duan, D.Li, B.Liu, Y.Ma, Z.Shao, H.Song, F.Tian, H.Xie, H.Yu. npj Comput. Mater., 5, 104(2019).
[40] C.Chen, P.Huang, X.Li, H.Liu, Y.Ma, Y.Sun, Y.Xie. J. Phys. Chem. Lett., 11, 935-939(2020).
[41] A.Bergara, G.Gao, X.Li, Y.Liu, G.Yang, X.Zhang. Phys. Rev. B, 105, 024504(2022).
[42] S. M.Clarke, H.-M.Huang, Y.Li, Y.-L.Li, E.Stavrou, Q.Zhu. Phys. Rev. B, 99, 220503(2019).
[43] X.Chen, D.Duan, Y.Liu, P.Lv, H.Sun, C.Wang. Solid State Commun., 293, 6-10(2019).
[44] J. A.Chevary, C.Fiolhais, K. A.Jackson, M. R.Pederson, J. P.Perdew, D. J.Singh, S. H.Vosko. Phys. Rev. B, 46, 6671-6687(1992).
[45] J.Furthmüller, G.Kresse. Phys. Rev. B, 54, 11169-11186(1996).
[46] P. E.Bl?chl. Phys. Rev. B, 50, 17953-17979(1994).
[47] A. D.Becke, K. E.Edgecombe. J. Chem. Phys., 92, 5397-5403(1990).
[48] V. L.Deringer, R.Dronskowski, C.Ertural, J.George, G.Hautier, R.Nelson. J. Comput. Chem., 41, 1931-1940(2020).
[49] F.Oba, I.Tanaka, A.Togo. Phys. Rev. B, 78, 134106(2008).
[50] S.Baroni, N.Bonini, M.Calandra, R.Car, C.Cavazzoni, D.Ceresoli, G. L.Chiarotti, M.Cococcioni, I.Dabo, A.Dal Corso, S.de Gironcoli, S.Fabris, G.Fratesi, R.Gebauer, U.Gerstmann, P.Giannozzi, C.Gougoussis, A.Kokalj, M.Lazzeri, L.Martin-Samos, N.Marzari, F.Mauri, R.Mazzarello, S.Paolini, A.Pasquarello, L.Paulatto, C.Sbraccia, S.Scandolo, G.Sclauzero, A. P.Seitsonen, A.Smogunov, P.Umari, R. M.Wentzcovitch. J. Phys.: Condens. Matter, 21, 395502(2009).
[51] O.Degtyareva, P.Dera, E.Gregoryanz, R. J.Hemley, H.-K.Mao, M.Somayazulu. Nat. Mater., 4, 152-155(2005).
[52] J. R.Van Wazer. J. Am. Chem. Soc., 78, 5709-5715(1956).
[53] T. K.Chattopadhyay, W.May, G. S.Pawley, H. G.von Schnering. Z. Kristallogr. - Cryst. Mater., 165, 47-64(1983).
[54] P. B.Allen, R. C.Dynes. Phys. Rev. B, 12, 905-922(1975).
[55] P. B.Allen, B.Mitrovi?. Solid State Phys., 37, 1-92(1983).
[56] J. P.Carbotte. Rev. Mod. Phys., 62, 1027(1990).
[57] Y.Ma, R. J.Needs, F.Peng, C. J.Pickard, Y.Sun, Q.Wu. Phys. Rev. Lett., 119, 107001(2017).
[58] V. Y.Fominski, A. G.Ivanova, A. G.Kvashnin, A. R.Oganov, V. M.Pudalov, A. V.Sadakov, D. V.Semenok, O. A.Sobolevskiy, V.Svitlyk, I. A.Troyan. Mater. Today, 33, 36-44(2020).
[59] W.Chen, T.Cui, D.Duan, X.Huang, X.Li, B.Liu, A. R.Oganov, D. V.Semenok, H.Xie, D.Zhou. Sci. Adv., 6, eaax6849(2020).
[60] T.Ishikawa, A.Nakanishi, K.Shimizu. J. Phys. Soc. Jpn., 87, 124711(2018).
[61] R.Arita, J. A.Flores-Livas, M.Hirayama, T.Koretsune, T.Nomoto, T.Wang. Phys. Rev. B, 104, 064510(2021).
[62] J.Hao, Y.Huang, W.Li, Y.Li, H.Liu, R. J.Needs, J. R.Nelson, C. J.Pickard, L.Wang, Y.Zhang. Phys. Rev. B, 93, 020103(2016).
[63] P. W.Anderson, P.Morel. Phys. Rev., 125, 1263-1271(1962).
[64] A.Davydov, K.Dewhurst, J. A.Flores-Livas, E. K. U.Gross, G.Profeta, A.Sanna, S.Sharma. J. Phys. Soc. Jpn., 87, 041012(2018).


Set citation alerts for the article
Please enter your email address

© Copyright 2018-2021 | Chinese Laser Press. All Rights Reserved 沪ICP备15018463号-20