
Qiaorui Gong, Chengchun Zhao, Yilun Yang, Qiannan Fang, Shanming Li, Min Xu, Yin Hang. Theoretical study on residual infrared absorption of Ti:sapphire laser crystals[J]. Photonics Research, 2021, 9(6): 909
- Photonics Research
- Vol. 9, Issue 6, 909 (2021)
Abstract
1. INTRODUCTION
Ti:sapphire is an important laser crystal and occupies a significant position in the super-intense ultrafast laser field due to its excellent laser characteristics (wide emission bandwidth, large peak power gain, and high damage threshold) and mature growth technology [1–4]. However, since people began to pay attention to the laser performance of Ti:sapphire crystal, the residual infrared absorption coinciding with the laser emission band has been a key problem affecting its laser efficiency [5–10]. Although it has been found that this absorption can be reduced to a certain extent by reduction annealing, it cannot be completely eliminated [5]. To solve this problem, the early experimental and theoretical works have considered and tried many approaches, such as sample preparation [5,9,11], an annealing process [5], a doping concentration [9], a defect mechanism and defect formation energy [12–19], and the crystal field environment [20]. These works did give some possible hypotheses and reasonable suggestions, among which the Ti ion pairs are the most controversial. The experimental research, however, is mostly based on the reverse deduction of phenomenon, while the theoretical research focuses on the exploration of defect type and defect energy. As a result, there was always a lack of direct evidence about the origin of the residual infrared absorption, especially in theory, that would correspond to the experimental phenomena. Therefore, it is necessary to systematically study the origin of this absorption.
Considering that traditional research methods are relatively macroscopic and strongly dependent on experimental conditions, it is not conducive to exploring the origin of material properties. The first principles method based on the density functional theory (DFT) has been proven to be an effective means to explore the physical and chemical properties of crystal materials and their micromechanisms. In this paper, by focusing on the energy-favorable defect models and models close to the real crystal environment, we defined four feasible Ti ion pair configurations according to the contact conditions of O octahedron in the crystal structure of . As a result, we have constructed four kinds of ion pair models and three kinds of ion pair models. The energy band structure, density of states (DOS), partial/local density of states (PDOS/LDOS), and the absorption spectra of Ti:sapphire crystals containing various defect models are calculated by the first principles method. We successfully obtained the absorption spectra consistent well with experiments in the line-contact ion pair and face-contact ion pair models and explained the mechanism from the energy level structure and electronic transition points of view. Through a comparison with the experimental phenomena and semi-quantitative analysis of their polarization characteristics, we are convinced that the residual infrared absorption peak at about 800 nm is jointly dominated by these two mechanisms. In addition, the charge distributions of the highest occupied molecule orbital (HOMO) and lowest unoccupied molecule orbital (LUMO) of the two models are calculated and analyzed, which provides a new perspective to reveal the nature of the residual infrared absorption. We believe that our theory of double mechanism dominant residual infrared absorption perfectly explains the annealing characteristics, polarization characteristics, and doping concentration dependence of the residual infrared absorption. Moreover, some other ion pair models provide a basis for understanding the more complex infrared absorption recently reported [9]. Our work, to the best of our knowledge, achieves the first breakthrough in theory for this key problem, which is of great significance for the development of Ti:sapphire laser crystals. We consider that this research can also provide a scheme to screen a new titanium-doped laser matrix with no residual infrared absorption.
2. THEORY AND CALCULATION
The basic starting point of the first principles method is to solve the Schrödinger equation of the system and obtain the wave function and energy eigenvalue of each state. However, this is almost impossible for a multi-electron system. The density functional theory (DFT) based on the Hohenberg–Kohn theorem [21] gives an available method, which uses the electron density distribution function of the system to replace the all-electron wave function. In this way, the solution of a system containing N electrons is reduced from 3N dimensions to three dimensions, which greatly reduces the difficulty of the calculation. The ground state energy and density of the multi-electron system can be obtained by solving the variational Kohn–Sham (K–S) [22] equation. After the ground state energy is determined, the ground state properties of the system can be predicted by taking various partial derivatives of the system energy. The Kohn–Sham equation is
Sign up for Photonics Research TOC. Get the latest issue of Photonics Research delivered right to you!Sign up now
The CASTEP code [23] based on the DFT and plane wave pseudopotential method is used for all calculations. The Perdew–Burke–Ernzerhof exchange-correlation potential adopts the generalized gradient approximation (GGA) [24]. Ultrasoft pseudopotentials [25] are applied for all atoms in the calculation. The plane-wave cutoff energy was set at 340 eV for all calculations. The Brillouin zone integration used the Monkhorst–Pack scheme [26] with a sampling mesh parameters for the k-point. The approximate separation between k-points used in energy band calculation is (1 Å = 0.1 nm). The k-point grid generated by the Monkhorst–Pack scheme is for the DOS and optical properties calculations. The energetic convergence threshold for the self-consistent field (SCF) is . During the geometry optimization process, the maximum energy change is , the maximum ionic displacement is , the max force is 0.03 eV/atom, and the max stress of the crystal is 0.05 GPa. The valence electronic configurations for pseudo atoms O, Al, and Ti are , , and , respectively.
3. RESULTS AND DISCUSSION
All the models we constructed are based on supercells () containing 120 atoms (72 O, 48 Al), and the coordination octahedrons geometry of the supercell and defect models are shown in Fig. 1. The optimized lattice parameters (, ) of the supercell are in good agreement with the experimental and theoretical data listed in Ref. [27]. Before discussing the results, there are some points to be demonstrated. First, limited by computing resources, although the supercells are used in our calculation, the atomic doping concentration of each model was still higher than that of the experiments. It does not, however, affect the qualitative analysis of the location and origin of each peak [27–33]. Ignoring the difference between the intensity of each peak and the experiments, we focus on the location of each absorption peak. Moreover, it is still meaningful to compare the absorption intensity among theoretical models to the same doping level. Second, the possible defects in real crystals are varied and interact with each other in a complex way. The absorption spectra in experiments often show the superposition of multiple mechanisms, which is difficult to be fully considered in a calculation. However, a specific model can be used to calculate and explain the possible effects of one defect or a class of defects in crystal with considerable accuracy. Third, the polarization absorption spectra of Ti:sapphire given in experiments tend to consider the -polarization () and -polarization (), but maybe only one direction of -polarization () is measured. This is feasible for real crystals with complex defect environments; however, in the theoretical model with only one defect, the influence of symmetry on each polarization direction satisfying may not be isotropic, which may cause the difference between the polarization strength of and given by theoretical calculations.
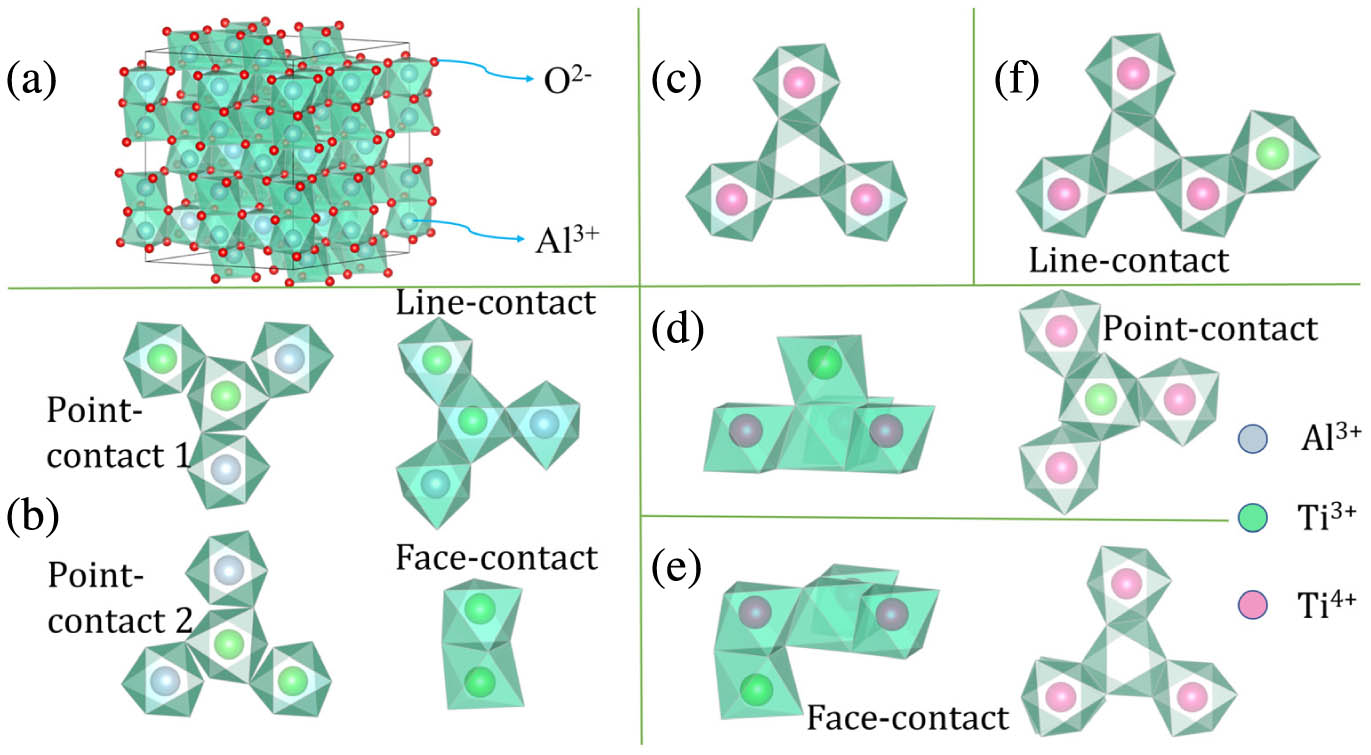
Figure 1.(a) Supercell structure of
Before discussing the calculation results of the ion pair models, we first investigate the absorption characteristics of a single substitutional titanium doping model. Since the contribution of substitutional to the main absorption (peak at 490 nm) has been confirmed by many experiments [5,7–9] and the crystal field theory of transition ions [20], the study of a single titanium doping model is helpful to test the feasibility and reliability of DFT in the study of a Ti:sapphire system. Figure 2 shows the simulated absorption spectra of a substitutional titanium model and the experimental absorption spectra of a certain Ti:sapphire sample grown by the heat exchange method. The calculation shows that there is a strong absorption peak near 485 nm, which is caused by the transition of a Ti_3d electron from to energy level with an energy of 2.54 eV. It is in good agreement with the absorption spectrum measured by the experiment, and matches well with the explanation given by the crystal field theory. This indicates that DFT is relatively reliable for the study of a Ti:sapphire system. In the simulated absorption spectrum of a substitutional titanium model, there is no residual infrared absorption band with a peak at 750–850 nm; however, in the measured absorption spectrum, there is a weak residual infrared absorption band near 790 nm.
![]()
Figure 2.Experimental absorption spectrum of Ti:sapphire sample with a doping concentration less than 0.16% (mass fraction) (on the left) and the simulated absorption spectrum of substitutional Ti-doped model with a theoretical doping concentration more than 3.6% (mass fraction) (on the right). There is a step at 860 nm on the experimental absorption spectrum, which is caused by the test system. The calculated PDOS distribution of Ti_3d is inserted into the simulated absorption spectrum.
Table 1 lists the calculation results of four ion pair models. As can be seen, the total energy of the line-contact model is the lowest. The total energy of each model does not simply increase with the increase of distance. It looks like the line-contact model is an anomaly. However, we think the reason for this anomaly is the face-contact model’s anomaly. Two ions in face-contact are too close to each other, which may cause core repulsion and large local distortion, making it more difficult for ion pairs in face-contact to form a binding model than it is for the line-contact with larger ion spacing. The analysis of the spin magnetic moment and spin polarization energy band structure indicates that, at a relatively close distance (face-contact, line-contact), the electrons of two ions tend to form antiferromagnetic coupling and share one d electron orbital; at a slightly larger distance (point-contact 2), the two electrons tend to be arranged in antiparallel spin and occupy two overlapped spin-polarized d orbitals, respectively, forming a ferrimagnetic coupling; at a larger distance (point-contact 1), the ferromagnetic coupling tends to form. The above analysis is consistent with the Heisenberg exchange interaction model for the origin of magnetism. According to the band gap value of the four ion pair models, the linear-contact model with antiferromagnetic coupling not only has the advantage in energy, but also a wider defect energy gap. It is in this model that the absorption spectrum with a high degree of consistency with the experimental results [5,7–10] is obtained.
| Distance (Optimized) | Magnetic Coupling | Band Gap | Total Energy | |
|---|---|---|---|---|
| Face-contact | 2.448 Å | Antiferromagnetic | 0.659 eV | −37453.6827 eV |
| Line-contact | 2.563 Å | Antiferromagnetic | 0.770 eV | −37453.8375 eV |
| Point-contact 2 | 3.034 Å | Ferrimagnetic | 0.543 eV | −37453.1967 eV |
| Point-contact 1 | 3.471 Å | Ferromagnetic | 0.299 eV | −37453.0835 eV |
Table 1. Main Parameters and Calculation Results for Four Kinds of
Figure 3 shows the band structure and absorption spectrum of the line-contact model. As shown in the band structure, the line-contact ion pair has a unique orbital energy level distribution near the Fermi level (0 eV), which leads to its unique absorption characteristics. There are three peaks in the absorption spectrum: 332 nm, 490 nm, and 799 nm. Combined with the analysis of the Ti_3d_PDOS and O_PDOS (the PDOS of O) shown in Fig. 4, the 490 nm main absorption and 799 nm near-infrared absorption are caused by the d-d transitions with energies of 2.53 eV and 1.54 eV, respectively. The near UV absorption at 332 nm is caused by the charge transfer transition from to O_2s2p, with an energy of 3.73 eV. It is worth noting that the polarization characteristics of the infrared absorption of this model are also consistent with that given in the experiments () [4,6], and the model also has a certain contribution to the main absorption in the visible region.
![]()
Figure 3.Band structure (on the left) and polarized absorption spectrum of line-contact
![]()
Figure 4.The DOS of the supercell
To further reveal the nature of the residual absorption induced by the line-contact ion pair model, we studied the charge distributions of HOMO and LUMO of this model. Figure 5 shows the calculated charge distributions of HOMO, LUMO, and (), as well as the corresponding energy intervals of the corresponding orbital distributions. It can be analogous to the width of an energy band near the Fermi surface in the energy band structure. HOMO corresponds to the first energy band under the Fermi level, while LUMO corresponds to the first energy band above the Fermi level. From the shape of charge distribution, the composition of these orbitals is mainly d orbitals, and the transitions from HOMO to LUMO () are all transitions. As can be seen, the charge distribution is basically concentrated around the two titanium ions in the various occupation cases. The light absorption we studied can be understood as that under the action of the incident photon electromagnetic field, the space charge gathered near the defect atom is redistributed, and this redistribution makes the energy of the system increase; that is, the system is excited. The transitions of electrons are mainly electric dipole transitions, and the transitions of transition ions belong to forbidden transitions, while some forbidden transitions can occur due to the action of the distorted crystal field. The HOMO charge distribution shown in Fig. 5(a) indicates that there is a large overlap region between the d orbitals of the two titanium ions, showing a bonding state with a lower energy. The larger blue-shaded area reflects the stronger electron delocalization ability and reactivity (the d electron is very localized, the delocalization here is only a relative expression). The energy difference between LUMO + 3, LUMO + 4, and HOMO is 1.54 eV and 2.53 eV, respectively, and the corresponding absorption wavelength is 805 nm and 488 nm, respectively, which matches well with the previous analysis based on the energy band and PDOS. In other words, under the action of incident photons with appropriate energy, the orbital charge distribution of the two Ti ions changes from the HOMO with lower energy to the LUMO + 3 or LUMO + 4 with higher energy, thus generating a certain intensity absorption of incident photons.
![]()
Figure 5.Optimized charge distributions of HOMO, LUMO, and
Moulton
The early works indicated that was a possible factor associated with the infrared absorption, and it was confirmed that the formation energy of this defect was almost the same as that of substitutional [13]. Inspired by the line-contact ion pair, we constructed one line-contact model [Fig. 1(c)], and the Al vacancy is considered as a charge compensation mechanism for ions. The calculation results shown in Fig. 6 indicate that the orbital energy levels of are higher than the Fermi level (0 eV), which conforms to the valence electron configuration of the ion ; in other words, there is no electron occupying in the d orbits. That means the transition in this model is an excited transition. On the one hand, this transition is less likely to occur; on the other hand, the absorption spectrum calculation does not include this part. Therefore, there is no main absorption or infrared absorption in the calculated absorption spectrum, but only absorption with a wavelength less than 300 nm. However, this part of the absorption involves the transition of the intrinsic energy level of the matrix, and its accuracy is still to be discussed. Because this paper does not focus on this part, it will not be elaborated. It can be determined that the model is not a direct contributor to the residual infrared absorption.
![]()
Figure 6.Band structure, Ti_3d_PDOS, and polarized absorption spectrum of
The ion pair model has been mentioned in some previous works [5,8,13] as a possible mechanism causing residual infrared absorption. However, there are so many possible configurations of this kind of defect models that no comprehensive report has been given in theory. Thus, based on the line-contact model, we constructed three kinds of ion pair models that are likely to cause residual infrared absorption. The results shown in Table 2 indicate that the line contact model has lower total energy, but the face-contact model has a larger defect energy gap. The absorption spectrum of the face-contact model is shown in Fig. 7, and there are two absorption peaks at 286 and 824 nm in this model. The analysis shows that the peak 286 nm is caused by the charge transfer transition from to , and the corresponding transition energy is 4.31 eV. Since this absorption is not the focus of this work, it will not be discussed further. It is noteworthy that the infrared absorption peak at 824 nm is consistent with the experimental absorption peak at about 750–850 nm [5,7–10]. The Ti_3d_PDOS in Fig. 7 shows that it is caused by the d–d transition with an energy of 1.50 eV. Figure 8 shows the charge distributions of HUMO, LUMO, and of the face-contact ion pair model. The detailed analysis can refer to the previous discussion on the line-contact model. Here, we mainly analyze the charge distributions of orbitals that relate to the residual absorption. The energy difference between and HOMO is about 1.49 eV, and the corresponding absorption wavelength is about 832 nm. According to the spatial charge distributions of the two situations shown in Figs. 8(a) and 8(f), their charge distributions are both concentrated on the face-contact Ti ion pair in the front of the figure, as shown in the geometry model in Fig. 1(e). The charge distribution of the two Ti ions of HOMO exists in a large overlapping region, while that of is concentrated around the two Ti ions separately, as if the bond is broken. Under the action of incident photons with suitable energy, such a transition of charge distribution from HOMO to is relatively likely to occur, thus generating a certain intensity of absorption to the incident light. Compared to the line-contact ion pair model mentioned above, we found that the face-contact ion pair model has a stronger near-infrared absorption at the same number of defects. This provides a reasonable explanation to understand the annealing absorption characteristics of Ti:sapphire. When annealed in a reducing atmosphere, the absorption caused by the face-contact ion pair will be largely eliminated. At this time, the residual infrared absorption is mainly contributed by the line-contact ion pair; the absorption will be reduced, but cannot be eliminated. When annealed in an oxidizing atmosphere, the residual infrared absorption is mainly contributed by the face-contact ion pair and the infrared absorption will be further enhanced.
| Band Gap | Total Energy | |
|---|---|---|
| Face-contact | 0.610 eV | −40487.4271 eV |
| Line-contact | 0.436 eV | −40487.5264 eV |
| Point-contact | 0.218 eV | −40487.3425 eV |
Table 2. Band Gap and Total Energy for Three Kinds of
![]()
Figure 7.Band structure, Ti_3d_PDOS, and polarized absorption spectrum of face-contact
![]()
Figure 8.Optimized charge distributions of HOMO, LUMO, and
We consider that the line-contact ion pair and face-contact ion pair are the two main mechanisms leading to the residual infrared absorption. As mentioned above, the absorption spectra (including the polarization characteristics) measured experimentally should be the result of the superposition of all mechanisms. To be self-consistent with the experimental phenomena, the superposition of polarization characteristics of the two mechanisms we obtained should satisfy . Thus, we make a semiquantitative analysis of the superposition of the two mechanisms. Suppose that there are line-contact ion pairs and face-contact ion pairs in one Ti:sapphire crystal. To satisfy , we need to satisfy . The calculation results indicate that . Thus, if , the polarization characteristics will be consistent with the experiments. In terms of the forming energy, the line-contact ion pair has the lowest forming energy (the largest energy advantage). Structurally, the ion pair involves four titanium ions and one aluminum vacancy, which will cause a large structural distortion and make it more difficult to form. Logically, the number of defects can satisfy . In addition, from the statistics of the experimental results, we found that the polarization strength of the residual infrared absorption at around 800 nm satisfies , and the value is not large. However, the line-contact ion pair satisfies and its , which is much larger than the experimental results. It means there should be another mechanism with strong polarization absorption at about 800 nm that satisfies , which just coincides with the polarization characteristic of the face-contact ion pair model. Because the face-contact ion pair only exists along the [001] direction, its absorption is mainly contributed by -polarization (see Fig. 7). In this way, the viewpoint that the residual infrared absorption is dominated by the double-mechanism forms a perfect self-consistency.
The discussions above only analyze the representative models, but other ion pair models provide a basis to explain more complex infrared absorption. For example, the infrared absorption at about 1130 nm is obtained in the face-contact ion pair model, the infrared absorption at 1000 nm is obtained in the line-contact ion pair model, and the polarized infrared absorption at 1390 nm is obtained in the point-contact ion pair model. In terms of energy, these models are not much larger than similar models, and thus it is possible they exist. In the research done by Moulton
4. CONCLUSIONS
In conclusion, this work provides a systematically theoretical study and direct theoretical evidence to explain the origin of the residual infrared absorption of Ti:sapphire laser crystals. For the first time, to the best of our knowledge, the near infrared absorption spectra consistent with experiments are obtained by theoretical calculation. The defect mechanism causing the residual infrared absorption is successfully determined. The theory that the residual infrared absorption was jointly dominated by the line contact ion pair and face contact ion pair was proposed, which successfully explains the annealing characteristics, doping concentration dependence, and polarization characteristics observed by experiments. In addition, the defect models that may lead to more complex residual infrared absorption are given. We believe, to the best of our knowledge, that we achieved a theoretical traceability and breakthrough for this key problem that affects the laser emission efficiency of Ti:sapphire crystals.
Acknowledgment
Acknowledgment. We acknowledge the support of researcher Zhi-Hua Yang from Xinjiang Key Laboratory of Electronic Information Materials and Devices, Xinjiang Technical Institute of Physics & Chemistry, Chinese Academy of Sciences.
References
[1] E. Cartlidge. The light fantastic. Science, 359, 382-385(2018).
[2] Z. Guo, L. H. Yu, J. Y. Wang, C. Wang, Y. Q. Liu, Z. B. Gan, W. Q. Li, Y. X. Leng, X. Y. Liang, R. X. Li. Improvement of the focusing ability by double deformable mirrors for 10-PW-level Ti:sapphire chirped pulse amplification laser system. Opt. Express, 26, 26776-26786(2018).
[3] W. Q. Li, Z. B. Gan, L. H. Yu, C. Wang, Y. Q. Liu, Z. Guo, L. Xu, M. Xu, Y. Hang, Y. Xu, J. Y. Wang, P. Huang, H. Cao, B. Yao, X. B. Zhang, L. R. Chen, Y. H. Tang, S. Li, X. Y. Liu, S. M. Li, M. Z. He, D. J. Yin, X. Y. Liang, Y. X. Leng, R. X. Li, Z. Z. Xu. 339 J high-energy Ti: sapphire chirped-pulse amplifier for 10 PW laser facility. Opt. Lett., 43, 5681-5684(2018).
[4] H. Cao, Z. B. Gan, X. Y. Liang, L. H. Yu, W. Q. Li, Z. Guo, P. Huang, J. Y. Wang, M. Xu, Y. Hang. Optical property measurements of 235 mm large-scale Ti:sapphire crystal. Chin. Opt. Lett., 16, 071401(2018).
[5] R. L. Aggarwal, A. Sanchez, M. M. Stuppi, R. E. Fahey, A. J. Strauss, W. R. Rapoport, C. P. Khattak. Residual infrared absorption in as-grown and annealed crystals of Ti:Al2O3. IEEE J. Quantum Electron., 24, 1003-1008(1988).
[6] B. Gu, M. Birnbaum, B. Leong, M. Bass. Material characteristics of titanium: sapphire. J. Opt. Soc. Am. B, 6, 2338-2341(1989).
[7] P. F. Moulton. Spectroscopic and laser characteristics of Ti:Al2O3. J. Opt. Soc. Am. B, 3, 125-133(1986).
[8] G. P. Zeng, S. T. Yin, X. L. Yu. The relation between infrared residual absorption and point defects in Ti3+:alpha-Al2O3 single crystal. Chin. J. Quantum Electron., 19, 25-30(2002).
[9] P. F. Moulton, J. G. Cederberg, K. T. Stevens, G. Foundos, M. Koselja, J. Preclikova. Characterization of absorption bands in Ti:sapphire crystals. Opt. Mater. Express, 9, 2216-2251(2019).
[10] I. V. Kryvonosov, L. A. Lytvynov. Properties of Ti-sapphire as laser material. Crystallogr. Rep., 57, 967-973(2012).
[11] G. Alombert-Goget, F. Trichard, H. Li, C. Pezzani, M. Silvestre, N. Barthalay, V. Motto-Ros, K. Lebbou. Titanium distribution profiles obtained by luminescence and LIBS measurements on Ti:Al2O3 grown by Czochralski and Kyropoulos techniques. Opt. Mater., 65, 28-32(2017).
[12] K. Matsunaga, A. Nakamura, T. Yamamoto, Y. Ikuhara. Theoretical study of defect structures in pure and titanium-doped alumina. Solid State Ion., 172, 155-158(2004).
[13] K. Matsunaga, A. Nakamura, T. Yamamoto, Y. Ikuhara. First-principles study of defect energetics in titanium-doped alumina. Phys. Rev. B, 68, 214102(2003).
[14] V. A. Pustovarov, T. V. Perevalov, V. A. Gritsenko, T. P. Smirnova, A. P. Yelisseyev. Oxygen vacancy in Al2O3: photoluminescence study and first-principle simulation. Thin Solid Films, 519, 6319-6322(2011).
[15] K. Matsunaga, T. Tanaka, T. Yamamoto, Y. Ikuhara. first-principles calculations of intrinsic defects in Al2O3. Phys. Rev. B, 68, 085110(2003).
[16] K. Matsunaga, T. Mizoguchi, A. Nakamura, T. Yamamoto, Y. Ikuhara. Formation of titanium-solute clusters in alumina: a first-principles study. Appl. Phys. Lett., 84, 4795-4797(2004).
[17] L. Y. Kravchenko, D. V. Fil. Defect complexes in Ti-doped sapphire: a first principles study. J. Appl. Phys., 123, 023104(2018).
[18] J. K. Bristow, D. Tiana, S. C. Parker, A. Walsh. Defect chemistry of Ti and Fe impurities and aggregates in Al2O3. J. Mater. Chem. A, 2, 6198-6208(2014).
[19] M. Yamaga, T. Yosida, S. Hara, N. Kodama, B. Henderson. Optical and electron spin resonance spectroscopy of Ti3+ and Ti4+ in Al2O3. J. Appl. Phys., 75, 1111-1117(1994).
[20] M. G. Brik, N. M. Avram, C. N. Avram. Exchange Charge Model of Crystal Field for 3d Ions, 36(2013).
[21] P. Hohenberg, W. Kohn. Inhomogeneous electron gas. Phys. Rev., 136, B864-B871(1964).
[22] L. J. Sham, W. Kohn. One-particle properties of an inhomogeneous interacting electron gas. Phys. Rev., 145, 561-571(1966).
[23] S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson, M. C. Payne. First principles methods using CASTEP. Z. Krist., 220, 567-570(2005).
[24] J. P. Perdew, K. Burke, M. Ernzerhof. Generalized gradient approximation made simple. Phys. Rev. Lett., 77, 3865-3868(1996).
[25] D. Vanderbilt. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B, 41, 7892-7895(1990).
[26] H. J. Monkhorst, J. D. Pack. Special points for Brillouin-zone integrations. Phys. Rev. B, 13, 5188-5192(1976).
[27] M. G. Brik.
[28] J. H. Zhang, J. W. Ding, Y. L. Zhang. Electronic and optical properties of Ti3+ doped α-Al2O3 crystals: first-principles calculations. Solid State Commun., 149, 1188-1192(2009).
[29] H. A. R. Alibad, S. R. Ghorbani. Structural and spin polarization effects of Cr, Fe and Ti elements on electronical properties of α–Al2O3 by first principle calculations. J. Mod. Phys., 02, 158-161(2011).
[30] H. Bao, X. L. Ruan. Absorption spectra and electron-vibration coupling of Ti:sapphire from first principles. J. Heat Transfer, 138, 042702(2016).
[31] A. F. Lima, J. M. Dantas, M. V. Lalic. An
[32] E. L. A. Eftimie, N. M. Avram. Absorption spectra, ligand field parameters and
[33] X. K. Hu, B. Wu, Y. Yang, Y. Y. Yeung, C. G. Ma, M. G. Brik. An old system revisited: Al2O3:Ti3+-microscopic crystal field effects explored by the crystal field and first-principles calculations. J. Alloys Compd., 847, 156459(2020).

Set citation alerts for the article
Please enter your email address
© Copyright 2018-2021 | Chinese Laser Press. All Rights Reserved 沪ICP备15018463号-20